Abstract
Aberrant nuclear accumulation of β-catenin is associated with cancer pathogenesis; however, the regulation of nuclear β-catenin was largely remained elusive. In J. Pathology, Paul et al. elucidated the molecular mechanism of accumulation of nuclear β-catenin in cancer [1].
Short Communication
β-Catenin is the central modulator of the canonical Wnt signaling pathway. Upon Wnt on state, β-Catenin is translocated to the nucleus and function as a transcription coactivator for several oncogenes. In Wnt off state, β-catenin is mostly localized in the cytoplasm and sequestered by the destruction complex, the negative regulator of β-catenin expression [2,3]. This destruction complex is composed of adenomatous polyposis coli (APC), casein kinase 1α (CK1α), glycogen synthase kinase 3α/β (GSK-3α/β), and AXIN1 [4-8]. CK1α initiates the degradation process of β-Catenin by phosphorylating it at Ser45, which is subsequently phosphorylated at Thr41, Ser37, and Ser33 by GSK3β [9]. The phosphorylated β-catenin is then polyubiquitinated by SCFβ-TrCP to promote its proteasomal degradation. In contrast, in the case of Wnt on state, β-catenin is released from the destruction complex and translocate to the nucleus. It is also reported that Norrin and R-Spondin activate Wnt signaling in stem cells and this activation is Wnt ligand independent [10-12]. They bind to LRP5/6 receptor and prevent the inactivation of LRP function by Rnf43/Znrf3. LRP5/6 functions as a coreceptor of Frizzled receptor and helps to separate Axin from the destruction complex. Thus, they help to translocate β-catenin in the nucleus through sequestration of Axin in the membrane. In the nucleus, β-catenin functions as a transcriptional coactivator to relate plethora of genes.
Destruction complex-mediated sequestration of β-catenin is efficient to prevent cancer. However, it is ineffective in most of the cancers because of either mutation in the components of the destruction complex [13,14] or in the β-Catenin itself [15,16]. Among the components of destruction complex, loss of APC is main driver for constitutive translocation of β-catenin in colorectal cancer [17]. APC is mutated in almost 49% of colorectal cancers [18]. However, β-Catenin is predominantly mutated in hepatocellular carcinoma, endometrial cancer, and pancreatic cancer [19-21]. In addition to APC inactivation and β-catenin mutation, mutation in R-spondin/LRP5/ RNF43 plays crucial role in Wnt-dependent colorectal cancer. RNF43 is mutated in approximately 19% of colorectal cancer. Similarly, R-spondin is mutated in 10% of colorectal cancer. Interestingly, irrespective of the type of breast cancer, canonical Wnt signaling is activated in almost 50% of breast cancer and is closely associated with reduced overall survival of the breast cancer patients [22]. It is important to note that in most of the breast cancer patients, expression of Wnt ligands and receptors are elevated, whereas antagonists are silence.
In addition to colorectal cancer and breast cancer, β-Catenin also plays an important role in generation of leukemia initiating cells from normal hematopoietic stem cells (HSC) as well as for self-renewal cells of leukemia initiating cells [23,24]. Further, β-Catenin acts as a major driver in T-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia [25,26]. BRAF(V600E) is the major driver in melanoma development and progression. Canonical Wnt signaling facilitates the BRAF/NRAS driven melanoma through preventing the oncogene induced senescence in melanocytes [27,28].
Nuclear localization of β-Catenin is very important for its oncogenic function. Therefore, several strategies have been developed to prevent the canonical Wnt signaling mediated cancer progression. Many of them are at different stages of clinical trials [18,29]. Several inhibitors were designed to inhibit the secretion of Wnt ligand through inhibition of Porcupine. In another strategy, small molecule inhibitors were developed to disrupt CBP and β-Catenin interaction. Small molecule inhibitors were also developed against the β-Catenin target genes to prevent cancer progression. In addition to small molecule inhibitor, antibody based therapy was also developed to inactivate frizzle receptors. However, many of these inhibitors showed severe side effects and some are at the next level of clinical trial. Recently, small molecule inhibitor was developed to promote the degradation of active form of β-Catenin [30]. However, small molecule-mediated proteasomal degradation of active form β-Catenin is not clear. Therefore, posttranslational regulation of nuclear β-Catenin was very important.
Recently, we have identified F-box protein FBXO16 as an important proteasomal regulator of nuclear β-Catenin [1]. FBXO16 is a relatively unexplored F-box protein and cellular function of FBXO16 was poorly understood. We therefore attempted to identify the cellular targets of FBXO16 through mass spectrometry study. We identified approximately 350 interactoms with minimum 3 unique peptides for each protein. Among these interactomes, β-Catenin was one. Further study revealed that FBXO16 attenuates the nuclear levels of β-Catenin cellular fractionation study. To support the attenuation of nuclear β-Catenin by FBXO16, transcriptional levels of β-Catenin regulated genes such as cyclin D1 and cMyc were examined. We also showed FBXO16-mediated regulation of β-Catenin using reporter assay. We found that mRNA levels of β-Catenin targeted genes were altered upon overexpression and depletion of FBXO16. To further support the observation, chromatin immunoprecipitation assay followed by RT-PCR analysis was performed to examine the recruitment of β-catenin on the promoter of Cyclin D1 as well as cMyc. Results revealed that recruitment of β-Catenin was impaired upon alteration of FBXO16 expression. We have validated our findings by both ectopic expression and ablation of FBXO16 in different cell lines. We found that the ablation of FBXO16 using multiple shRNAs leads to enhanced binding of β-catenin to the promoter of cyclin D1 and cMyc followed by their heightened transcriptional levels.
Previous studies have identified two different modes of Wnt-β-catenin signaling pathway activation, via Wnt ligand activation and non-Wnt ligand-mediated activation [31-33]. Our findings showed that FBXO16 promotes the degradation of nuclear β-Catenin even in Wnt activated condition. For example, FBXO16 efficiently directs the degradation of β-Catenin in the presence of Wnt3a ligand as well as epidermal growth factor (EGF), the activators of the Wnt signaling pathway. These findings were well supported by conventional TOP/FOP assay. We showed that despite the presence of canonical Wnt activation signals like EGF and Wnt3a, ectopic presence of FBXO16 leads to reduction of luciferase activity. In contrast, depletion of FBXO16 results in significant augmentation of luciferase activity.
Many of the transcriptional target genes of β-catenin are well known oncogenes and have an established role in the initiation, establishment, and progression of cancer [33]. We therefore monitored epithelial to mesenchymal (EMT) transitions, a process known to play a role in oncogenesis, to assess the breast cancer malignancy [34]. Previous studies have shown that Snail and Twist enhance the aggressiveness of cancer cells through augmenting EMT process [34,35]. Snail and Twist are the important targets of β-Catenin. Indeed, we found that FBXO16 promotes the expression of E-cadherin and restricts the expression of oncogenic factors such as cMyc, Cyclin D1, Snail, and Twist. Further investigation revealed that FBXO16 controls the expression levels of these EMT regulators by monitoring the cellular levels of β-Catenin.
Human genome encodes genes for 69 F-box proteins and cellular function of majority of the F-box proteins are yet to be discovered. FBXO16 was one of the uncharacterized F-box proteins. Cellular function as well as its targets was not known. Our study, for the first time, systematically discovered the cellular function as well as cellular targets of FBXO16. F-box proteins are the integral component of SCF (SKP1-Cullin1-F-box protein) ubiquitin ligase complex. SCF complex consists of three invariable components (SKP1, Cullin1 and RBX1) and F-box protein as variable components. F-box proteins assembled in the SCF complex through utilizing their “F-box” motif, a 40 – 50 amino acid conserved motif. F-box proteins recognize mostly phosphorylated substrates and bring them to the SCF complex to ubiquitinate. Since F-box protein can target multiple cellular proteins, we identified different interacting partners of FBXO16 using mass spectrometrybased proteomic study. Our mass spec analysis revealed more than 350 interacting partners. In addition, β-Catenin, we have identified as another important oncogene clathrin heavy chain (CLTC). CLTC is known to play a pivotal role in vesicular trafficking and post-mitotic Golgi reassembly [36]. Further, pathway analysis of its interactomes revealed its association with various cellular signaling pathways, biological regulatory networks, metabolism, cellular trafficking, and immune response (Figure 1). Given the diverse interactomes of FBXO16, it will be interesting to understand its role in different cellular processes in future. We believe this study has laid the base by establishing one critical function of FBXO16, however, more of its important functions are yet to be deciphered.
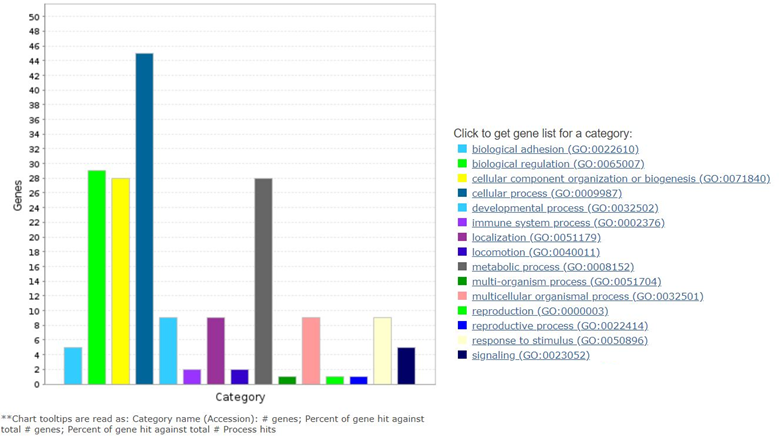
Figure 1. Analysis of FBXO16 interactome’s association with diverse cellular pathways analyzed by PANTHER.
Most of the F box protein associates with the scaffolding protein of SCF complex through the F-box domain. To further characterize whether FBXO16 also forms SCF complex to target β-catenin degradation, we generated F-box motif deleted FBXO16 mutant. The mutant is incompetent to degrade nuclear β-catenin solely due to its inability to form SCF complex. Notably, most of cancer reports revealed an aberrant activation of the Wnt signaling pathway leading to accumulation of β-catenin and its downstream pathway activation [37,33]. Interestingly, we found that the FBXO16-mediated degradation of nuclear β-catenin is independent of activation status of Wnt, which we think is quite significant in terms of development of therapy. There is a nice inverse expression relationship of FBXO16 and nuclear β-catenin in human breast cancer patient samples. Our study also sheds a light on the topology of FBXO16. Analysis of the sequence of FBXO16 revealed the presence of a nuclear localization signal (NLS) in the C-terminal of FBXO16 (247-273 amino acid region). We found that this NLS signal is essential for the FBXO16 mediated regulation of nuclear β-Catenin, and depletion of this NLS leads to loss of nuclear localization of FBXO16 as well as attenuates its ability to regulate the levels of nuclear β-Catenin. Therefore, inactivation of pathway(s) responsible for repression of FBXO16 expression in cancer could be an important strategy to prevent nuclear accumulation of β-Catenin and hence cancer progression. Although we found that FBXO16 contains an NLS and it shuttles between cytoplasmic and nuclear compartment, it is still unclear why FBXO16 shuttle between two cellular compartments and what are the controlling factors. Future studies directed towards this direction can make significant advancement in understanding FBXO16’s function.
Chemotherapy is the first line of treatment for almost all cancers. However, resistance towards chemotherapeutic drugs is the biggest hurdles physicians encounter during chemotherapy. Earlier report has shown the reduced sensitivity of MDA-MB-231 breast cancer cell lines towards chemotherapeutic agent Doxorubicin [38]. Recently, we demonstrated that FBXO16 functions as a putative tumor suppressor through attenuating β-catenin and has undetectable expression in MDA-MB-231cell line. We therefore investigated whether FBXO16 could be an adjuvant during chemotherapy in triple negative breast cancer cell line MDA-MB-231. Interestingly, we observed that treatment of Doxorubicin alone or only overexpression of FBXO16 does not have any significant growth suppressive effect on MDA-MB-231. However, ectopic expression of FBXO16 during Doxorubicin treatment results in three-fold increase apoptotic cell death of MDAMB- 231 cells. Therefore, it will be interesting to explore the therapeutic potential of FBXO16 in breast cancer as well as other cancers alone or in combination with different chemotherapeutic, radiation, and immunotherapy.
In our recent study, we have demonstrated cellular function of FBXO16, an unexplored F-box protein. We have found a wide array of its interacting partners including nuclear β-Catenin as a crucial substrate. Our study established FBXO16 as a putative tumor suppressor gene. Our detailed investigation has revealed the expression profile of FBXO16 in different human breast cancer cell lines as well as breast cancer patient samples. FBXO16 controls different cellular processes such as cell proliferation, migration, survival and tumor formation through attenuating cellular function of nuclear β-Catenin. Ubiquitin dependent degradation of β-Catenin by FBXO16 is unique in many aspects from other β-Catenin regulators. For instance, well known F-box protein β-TrCP1 regulates cytoplasmic pool of β-Catenin whereas F-box protein FBXO16 regulates nuclear β-Catenin. β-TrCP1-mediated β-Catenin degradation is GSK3β kinase activity dependent whereas FBXO16 directs the degradation of nuclear β-Catenin in GSK3β independent manner. Huang and colleagues have shown that nuclear β-Catenin could be degraded by TRIM33 [39]. However, TRIM33-mediated degradation of nuclear β-Catenin is PKCδ dependent. However, nuclear localization of PKCδ is also signal-dependent. Thus, degradation of nuclear β-Catenin by FBXO16 is also distinct from TRIM33. TRIM33 directs degradation of nuclear β-Catenin in PKCδ kinase dependent manner whereas FBXO16 directs nuclear β-Catenin degradation in PKCδ independent manner. Moreover, we showed that FBXO16 could attenuate nuclear β-Catenin even in the presence of Wnt ligand and EGF. However, expression of FBXO16 is compromised in most of the higher grade of cancers. Recent study showed that FBXO16 is located in loss of heterozygosity of chromosomal region 8p21.1 in breast cancer [40]. However, we are not ruling out the involvement of other mechanisms such as epigenetic silencing, mutations, and proteasomal regulation for the compromised expression of FBXO16 in different cancers. Additionally, it will be interesting to understand the role of nuclear envelop proteins and nuclear pore complexes that controls the translocation of proteins like β-Catenin [41,42]. Therefore, based on recent studies, we speculate that utilizing FBXO16 to target nuclear β-Catenin could be an effective therapeutic alternative. A recent study has identified molecular glue that enhances the degradation of β-Catenin via β-TrCP [43]. Though it is a significant pharmacological enhancement, but it might not be effective to control the nuclear accumulation of enhanced nuclear β-Catenin accumulation and wnt signal activation in cancer. Therefore, we think a similar approach can be taken to identify molecular glues or even PROTACS which may selectively enhance the interaction of FBXO16 and β-Catenin or selectively target nuclear β-Catenin degradation by FBXO16 (Figure 2).
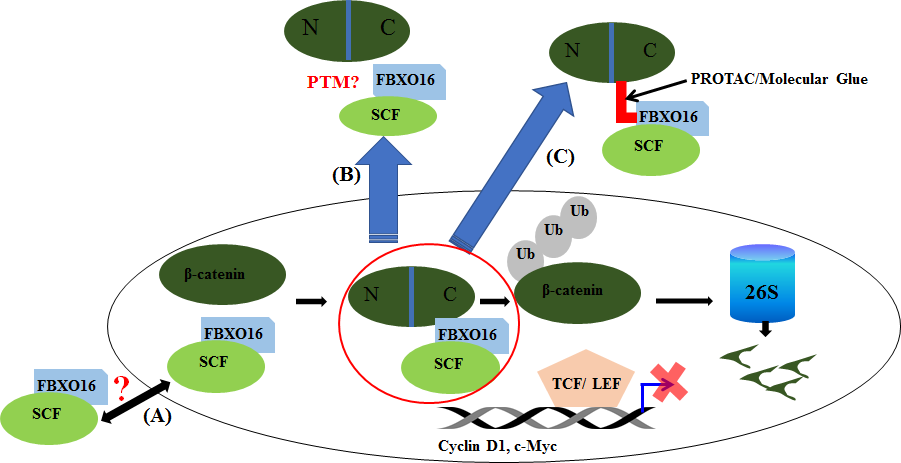
Figure 2. Model describes FBXO16-mediated regulation of nuclear β-Catenin., The prospective: FBXO16 inhibits the oncogenic activity of nuclear β-Catenin by promoting its proteasomal degradation. In future, it will be interesting to address whether FBXO16 shuttles between nucleus and cytoplasm (A), involvement of posttranslational modification (PTM) in FBXO16 in the shuttling process as well as interaction with nuclear β-Catenin (B) and development of PROTACs/Molecular glues to facilitate ablation of nuclear β-Catenin through proteasome mediated degradation (C).
Declaration
Authors declare that we do not have any competing financial interests in relation to the work described here.
Acknowledgement
This work was supported in part by National Centre for Cell Science, Department of Biotechnology, Ministry of Science and Technology, Government of India (to M.K.S.).
References
2. Martin GS. Cell signaling and cancer. Cancer cell. 2003 Sep 1;4(3):167-74.
3. Lin SY, Xia W, Wang JC, Kwong KY, Spohn B, Wen Y, et al. β- catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proceedings of the National Academy of Sciences. 2000 Apr 11;97(8):4262-6.
4. Li VS, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP, et al. Wnt signaling through inhibition of β-catenin degradation in an intact Axin1 complex. Cell. 2012 Jun 8;149(6):1245-56.
5. Gao ZH, Seeling JM, Hill V, Yochum A, Virshup DM. Casein kinase I phosphorylates and destabilizes the β-catenin degradation complex. Proceedings of the National Academy of Sciences. 2002 Feb 5;99(3):1182-7.
6. Ha NC, Tonozuka T, Stamos JL, Choi HJ, Weis WI. Mechanism of phosphorylation- dependent binding of APC to β-catenin and its role in β-catenin degradation. Molecular Cell. 2004 Aug 27;15(4):511-21.
7. Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, et al. Control of β- catenin phosphorylation/ degradation by a dual-kinase mechanism. Cell. 2002 Mar 22;108(6):837-47.
8. Wu G, Huang H, Abreu JG, He X. Inhibition of GSK3 phosphorylation of β-catenin via phosphorylated PPPSPXS motifs of Wnt coreceptor LRP6. PloS One. 2009 Mar 18;4(3):e4926.
9. Hagen T, Vidal-Puig A. Characterisation of the phosphorylation of β-catenin at the GSK-3 priming site Ser45. Biochemical and Biophysical Research Communications. 2002 Jun 7;294(2):324-8.
10. Xu Q, Wang Y, Dabdoub A, Smallwood PM, Williams J, Woods C, et al. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004 Mar 19;116(6):883-95.
11. Kazanskaya O, Glinka A, del Barco Barrantes I, Stannek P, Niehrs C, Wu W. R- Spondin2 is a secreted activator of Wnt/β-catenin signaling and is required for Xenopus myogenesis. Developmental Cell. 2004 Oct 1;7(4):525-34.
12. de Lau W, Peng WC, Gros P, Clevers H. The R-spondin/ Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes & Development. 2014 Feb 15;28(4):305-16.
13. Mazzoni SM, Fearon ER. AXIN1 and AXIN2 variants in gastrointestinal cancers. Cancer Letters. 2014 Dec 1;355(1):1-8.
14. Kwong LN, Dove WF. APC and its modifiers in colon cancer. In:Apc Proteins. Springer: New York, NY. 2009: pp. 85-106.
15. Machin P, Catasus L, Pons C, Muñoz J, Matias-Guiu X, Prat J. CTNNB1 mutations and β-catenin expression in endometrial carcinomas. Human pathology. 2002 Feb 1;33(2):206-12.
16. Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al.Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science. 1997 Mar 21;275(5307):1787-90.
17. Polakis P. Wnt signaling in cancer. Cold Spring Harbor Perspectives in Biology. 2012 May 1;4(5):a008052.
18. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017 Mar;36(11):1461-73.
19. Grigoryan T, Wend P, Klaus A, Birchmeier W. Deciphering the function of canonical Wnt signals in development and disease: conditional loss-and gainof- function mutations of β-catenin in mice. Genes & Development. 2008 Sep 1;22(17):2308-41.
20. Harada N, Tamai Y, Ishikawa TO, Sauer B, Takaku K, Oshima M, et al. Intestinal polyposis in mice with a dominant stable mutation of the β-catenin gene. The EMBO Journal. 1999 Nov 1;18(21):5931-42.
21. Kim S, Jeong S. Mutation hotspots in the β-catenin gene: lessons from the human cancer genome databases. Molecules and Cells. 2019 Jan 31;42(1):8.
22. Lin SY, Xia W, Wang JC, Kwong KY, Spohn B, Wen Y, et al. β- catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proceedings of the National Academy of Sciences. 2000 Apr 11;97(8):4262-6.
23. Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, et al. The Wnt/β-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010 Mar 26;327(5973):1650-3.
24. Yeung J, Esposito MT, Gandillet A, Zeisig BB, Griessinger E, Bonnet D, So CW. β- Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell. 2010 Dec 14;18(6):606-18.
25. Giambra V, Jenkins CE, Lam SH, Hoofd C, Belmonte M, Wang X, et al. Leukemia stem cells in T-ALL require active Hif1α and Wnt signaling. Blood, The Journal of the American Society of Hematology. 2015 Jun 18;125(25):3917- 27.
26. Lu D, Zhao Y, Tawatao R, Cottam HB, Sen M, Leoni LM, et al. Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proceedings of the National Academy of Sciences. 2004 Mar 2;101(9):3118-23.
27. Juan J, Muraguchi T, Iezza G, Sears RC, McMahon M. Diminished WNT→ β-catenin→ c-MYC signaling is a barrier for malignant progression of BRAFV600Einduced lung tumors. Genes & Development. 2014 Mar 15;28(6):561-75.
28. Delmas V, Beermann F, Martinozzi S, Carreira S, Ackermann J, Kumasaka M, et al. β-Catenin induces immortalization of melanocytes by suppressing p16INK4a expression and cooperates with N-Ras in melanoma development. Genes & Development. 2007 Nov 15;21(22):2923-35.
29. Jung YS, Park JI. Wnt signaling in cancer: therapeutic targeting of Wnt signaling beyond β-catenin and the destruction complex. Experimental & Molecular Medicine. 2020 Feb 10:1-9.
30. Hwang SY, Deng X, Byun S, Lee C, Lee SJ, Suh H, et al. Direct targeting of β-catenin by a small molecule stimulates proteasomal degradation and suppresses oncogenic Wnt/β-catenin signaling. Cell Reports. 2016 Jun 28;16(1):28-36.
31. Ackers I, Malgor R. Interrelationship of canonical and non-canonical Wnt signalling pathways in chronic metabolic diseases. Diabetes and Vascular Disease Research. 2018 Jan;15(1):3-13.
32. Valenta T, Hausmann G, Basler K. The many faces and functions of β-catenin. The EMBO Journal. 2012 Jun 13;31(12):2714-36.
33. Shang S, Hua F, Hu ZW. The regulation of β-catenin activity and function in cancer: therapeutic opportunities. Oncotarget. 2017 May 16;8(20):33972.
34. Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nature Reviews Cancer. 2018 Feb;18(2):128.
35. Martin TA, Goyal A, Watkins G, Jiang WG. Expression of the transcription factors snail, slug, and twist and their clinical significance in human breast cancer. Annals of Surgical Oncology. 2005 Jun 1;12(6):488-96.
36. Radulescu AE, Shields D. Clathrin is required for postmitotic Golgi reassembly. The FASEB Journal. 2012 Jan;26(1):129-36.
37. Khramtsov AI, Khramtsova GF, Tretiakova M, Huo D, Olopade OI, Goss KH. Wnt/β- catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. The American Journal of Pathology. 2010 Jun 1;176(6):2911-20.
38. Sapio L, Sorvillo L, Illiano M, Chiosi E, Spina A, Naviglio S. Inorganic phosphate prevents Erk1/2 and Stat3 activation and improves sensitivity to doxorubicin of MDA- MB-231 breast cancer cells. Molecules. 2015 Sep;20(9):15910-28.
39. Xue J, Chen Y, Wu Y, Wang Z, Zhou A, Zhang S, et al. Tumour suppressor TRIM33 targets nuclear β-catenin degradation. Nature Communications. 2015 Feb 2;6(1):1- 6.
40. Cai Y, Crowther J, Pastor T, Asbagh LA, Baietti MF, De Troyer M, et al. Loss of chromosome 8p governs tumor progression and drug response by altering lipid metabolism. Cancer Cell. 2016 May 9;29(5):751-66.
41. Chow KH, Factor RE, Ullman KS. The nuclear envelope environment and its cancer connections. Nature Reviews Cancer. 2012 Mar;12(3):196-209.
42. Alvarado-Kristensson M, Rosselló CA. The biology of the nuclear envelope and its implications in cancer biology. International Journal of Molecular Sciences. 2019 Jan;20(10):2586.
43. Simonetta KR, Taygerly J, Boyle K, Basham SE, Padovani C, Lou Y, et al. Prospective discovery of small molecule enhancers of an E3 ligase-substrate interaction. Nature Communications. 2019 Mar 29;10(1):1-2.