Abstract
Background: Anti-N-methyl-D-aspartate receptor encephalitis (anti-NMDARE) is one of the most prevalent forms of autoimmune encephalitis in the pediatric population. However, its diagnosis remains challenging due to the broad spectrum of clinical manifestations (psychiatric symptoms and seizures most typically) and mainly normal neuroimaging findings. Recent literature has highlighted an association with inflammatory demyelinating disorders, further complicating the differential diagnosis.
Case Description: A 13-year-old girl presented with new-onset epileptic seizures, behavioral disturbances and MRI abnormalities suggestive of a demyelinating disease involving both the brain and spinal cord. Immunosuppressive treatment was initiated, consisting of high-dose corticosteroids and plasma exchange, followed by second-line treatment with rituximab. Anti-N-methyl-D-aspartate receptor (anti-NMDAR) antibodies were detected in the cerebrospinal fluid (CSF), while all other tested antibodies, including MOG IgG (Myelin Oligodendrocyte Glycoprotein) and AQP4 IgG (Aquaporin-4), were negative in both CSF and serum. Eight months later, the patient experienced a clinical relapse, which responded favorably to repeated rituximab administration. Notably, no symptoms or signs compatible with demyelinating disease were observed throughout the disease course.
Conclusion: Overlapping syndromes involving anti-NMDARE, multiple sclerosis (MS), MOG antibody-associated disease (MOGAD) and AQP4-antibody-negative neuromyelitis optica spectrum disorder (NMOSD) are increasingly recognized in clinical practice. Although these overlap syndromes remain uncommon in the pediatric population, they represent an important diagnostic consideration due to their implications for treatment and prognosis. Spinal cord abnormalities, as observed in this case of an anti-NMDARE-MS overlap syndrome, are particularly rare and underscore the complexity of diagnosing and treating primary autoimmune encephalitis with coexisting demyelinating disorders.
Keywords
Anti-N-methyl-D-aspartate receptor encephalitis, Demyelinating diseases, Overlap syndromes, Spinal lesion
Introduction
Anti-N-methyl-D-aspartate receptor encephalitis (anti-NMDARE) is an autoantibody mediated neurological disorder first identified in 2007 [1]. Today, anti-NMDARE is known as one of the most prevalent forms of non-viral encephalitis, particularly in children [2]. Autoantibodies target the glutamate receptor N1 (GluN1) subunit of the NMDA receptor (NMDAR) resulting in internalization of the NMDAR. This receptor plays a role in synaptic plasticity and mediating the process of learning, memory function, cognition and perception of sensory stimuli [3,4]. GluN1 is predominantly expressed in the frontotemporal regions of the brain [5]. Up to 50% of patients may present with a normal MRI (Magnetic Resonance Imaging) [6]. More and more evidence suggests an association between anti-NMDARE and co-existing or sequential demyelinating disorders. In such cases, MRI abnormalities are typically more extensive or multifocal compared to those observed in patients with isolated anti-NMDARE [7].
The clinical presentation of anti-NMDARE is highly variable and characterized by neuropsychiatric symptoms that can make the diagnosis challenging. The disorder predominantly affects young females (mostly age under 21 years) with a female-to-male ratio of 4:1. In post-pubertal women, approximately 50% of anti-NMDARE cases are associated with an ovarian teratoma, whereas the incidence drops to 9% in pre-pubertal females. Early diagnosis and treatment are significant prognostic factors, making timely initiation of appropriate immunotherapy and complete tumor resection in paraneoplastic cases critically important [5]. Most children with anti-NMDARE show good recovery; however, nearly 20% experience long term behavioural difficulties or learning problems [8]. Recent studies show an association of extensive white matter lesions and level of cognitive dysfunction, including memory and executive deficits [9,10]. Abnormal MRI at disease onset is linked with worse clinical outcome [11].
Here, we present a case of a female adolescent exhibiting clinical symptoms consistent with anti-NMDARE, accompanied by central imaging findings suggestive of a demyelinating disease. This represents a rare observation, highlighting the importance of recognizing that anti-NMDARE can co-exist with other central nervous system inflammatory disorders.
Case Presentation
A 13-year-old post-pubertal girl with no significant prior history, presented to the emergency department after a nocturnal self-limiting episode of breathing difficulties and rhythmic body movements. The patient had no recent history of sickness or trauma. Upon evaluation in the emergency department, the patient developed several short tonic-clonic seizures. Comprehensive work-up including blood analysis, urine toxicology, and brain computed tomography (CT) scan revealed no abnormalities. The patient was admitted to the hospital for observation, but a few days later discharged without further incidents.
However, a scheduled MRI of the brain (without gadolinium administration) a few days later revealed multiple supratentorial FLAIR/T2 hyperintense white matter lesions. These lesions were located periventricular, juxtacortical or on the calloseptal interface and/or had an orientation perpendicular to the lateral ventricles, raising suspicion of multiple sclerosis (Figures 1a and 1b). There was no central vein sign observed. The MRI of the spinal cord was inconclusive due to movement artefacts, but there was a suspicion of small T2 hyperintense intramedullary lesions at levels C2 and T11 (Figure 1c).
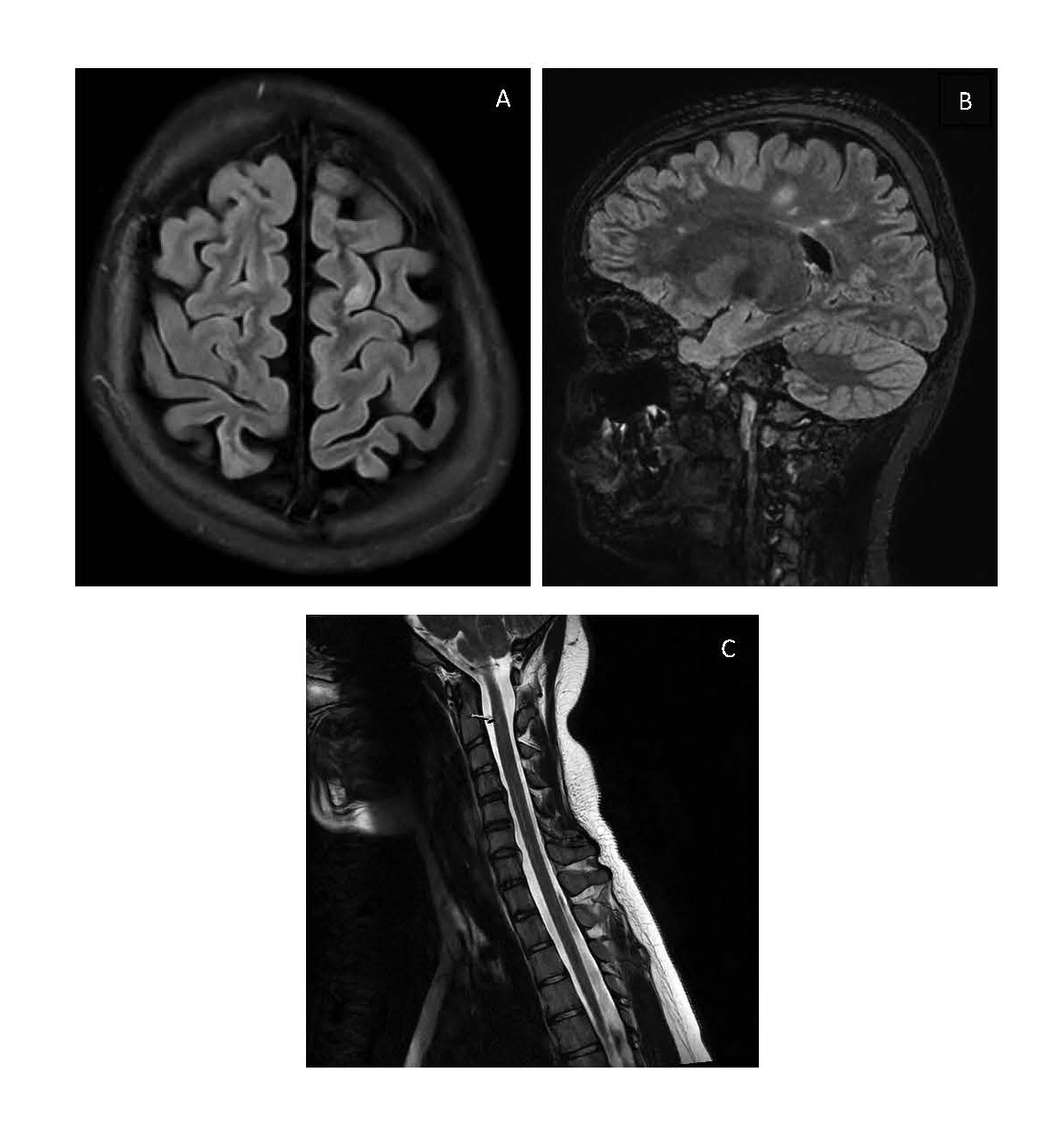
At that moment, the patient’s mother reported her daughter to present behavioural changes, including memory deficits, apathy, and automatisms (smacking movements, repeatedly sliding chair under the table). Cerebrospinal fluid (CSF) analysis showed no pleocytosis (only 4 white blood cells (WBC)) and normal protein (41mg/dl) but revealed an elevated immunoglobulin G (IgG) index of 5.8 (normal value 0.30-0.60). Oligoclonal bands were not analyzed. Ophthalmic evaluation with fundoscopy and Optical Coherence Tomography (OCT) was normal.
Given the presumption of an autoimmune encephalitis, the patient was readmitted and initiated on high-dose intravenous corticosteroids (30 mg/kg with a maximum of 1 gram methylprednisolone once a day for five days). Four days later, she was transferred to the paediatric intensive care unit because of clinical deterioration. The patient exhibited a waxing and waning pattern of catatonia, with prominent features including areactivity, mutism, and posturing. There were no signs of autonomic dysfunction. Additionally, she developed focal epileptic seizures for which intravenous midazolam and levetiracetam were administered with complete resolution of seizures. Repeat 3T MRI with IV contrast administration, showed a similar pattern to prior studies with 2 new white matter lesions in the left temporal and right frontal lobe, decrease in size of one lesion in the left frontal lobe and confirmation of the intramedullary lesions. No contrast enhancing lesions were seen. After five days of high-dose corticosteroid treatment without clinical improvement, seven sessions of plasma-exchange (PLEX) were initiated. Meanwhile, CSF analysis returned positive for anti-NMDAR antibodies (fixed cell-based assay, Euroimmun®), while serum antibody tests (anti-NMDAR, anti-myelin oligodendrocyte glycoprotein (anti-MOG) and anti-aquaporin-4 (anti-AQP4)) remained negative. MRI of the pelvis could not demonstrate an ovarian teratoma.
Despite PLEX and corticosteroids, clinical improvement was inadequate and second-line immunotherapy was initiated with weekly administrations of rituximab (375 mg/m2). After four doses, the patient showed significant clinical improvement, becoming more cooperative and gradually regaining speech and self-care abilities. She was subsequently enrolled in a multidisciplinary rehabilitation program where she quickly regained her pre-illness level of functioning. Brain MRI four months after clinical onset, showed no new lesions and a mild reduction in the size of multiple supratentorial white matter lesions. Repeated electroencephalography (EEG) was normal, so levetiracetam was gradually reduced and stopped.
However, three months later, she presented again at our emergency department with convulsions. A relapse of anti-NMDARE was suspected and a new lumbar puncture and brain MRI were performed. CSF analysis showed 27 WBC (mostly mononuclear cells), negative viral PCR and bacterial culture, but again an elevated IgG index (3.09). Approximately 10 oligoclonal bands were detected only in the CSF. Anti-NMDAR antibodies were found in serum and liquor, other antibodies remained negative, including anti-MOG en anti-AQP4. Follow-up brain MRI showed no new lesions and again a subtle reduction in size of some of the white matter lesions. Treatment with high-dose corticosteroids (30 mg/kg with a maximum of 1 gram methylprednisolone once a day for five days) was initiated leading to normalization of behavior within the following days, without new epileptic events occurring. Flowcytometry showed that there was incomplete B-cell depletion (100 CD19+ B-cells/mcL) after prior induction with rituximab. Therefore, it was decided to restart rituximab, 1 gram with a repeat dose after 2 weeks and then after 6 months. Repeated MRI pelvis showed no teratoma. Again, she regained her pre-illness level of functioning and showed no neurological deficits. Repeated MRI of the spinal cord, performed one year after initial diagnosis, demonstrated a reduction to complete resolution of the hyperintense lesions, with no additional abnormalities observed.
Discussion
Even with the current understanding of anti-NMDARE, accurate and prompt diagnosis of the disease remains challenging. Our case illustrates rare findings in a pediatric patient with anti-NMDARE and concomitant asymptomatic demyelinating lesions, fulfilling 2024 McDonald diagnostic criteria for multiple sclerosis, and therefore this case is an example of an NMDARE-MS overlap syndrome. The clinical symptoms of our patient were consistent with anti-NMDARE but not with a demyelinating pathology. The phenomenon of radiologic findings suggestive of demyelinating pathology without clinical symptoms is referred to as Radiologically Isolated Syndrome (RIS). It has been demonstrated that the majority of patients with RIS develop clinical symptoms of multiple sclerosis (MS) within a ten-year period [12]. Close clinical and radiological follow-up for development of symptoms associated with MS is warranted. Whether early treatment with a highly efficacious disease modifying therapy such as rituximab would delay or even abort the onset of clinical MS symptoms remains to be investigated.
There are a few case reports of demyelinated lesions resembling MS in cases of adults with anti-NMDARE [13,14], but a literature search demonstrated only one other pediatric case. In this other case report, a girl first presented with symptoms of inflammatory demyelinating disease of the CNS and positive anti-NMDAR antibodies, followed by anti-NMDAR encephalitis symptoms, and finally diagnosed with MS [15].
There are more and more studies regarding white matter alterations, found by advanced MRI techniques like DTI (Diffusion Tensor Imaging), in anti-NMDARE patients. These can provide important information for prognosis, since conventional MRI is unremarkable or non-specific in most patients. Yang et al. make the hypothesis that these white matter alterations are the result of demyelination, by interpretation of the different metrics of MRI DTI technique [16].
NMDARE and MS overlap syndromes have been recently recognized [17], while also cases of NMDARE followed by MOG antibody-associated disease (MOGAD) or AQP4-antibody-negative neuromyelitis optica spectrum disorder (NMSOD) have been described [18]. Alternatively, a simultaneous presentation or initial NMSOD of MOGAD followed by anti-NMDARE is also reported and defined as MOGAD and NMDARE overlapping syndrome (MNOS) [18]. The coexistence of multiple auto-antibody related diseases is increasingly recognized, though an etiological explanation remains elusive [19]. It is unclear whether the antibodies against neuronal receptors cause demyelination or do they develop as a consequence of it? [20]. Previous studies have shown that NMDARs are present on the myelin sheath formed by oligodendrocytes, which suggests that myelin injury resulting from a pathological burst of anti-NMDAR antibodies may activate or exacerbate inflammation in multiple sclerosis (MS) [15]. It is notable that the occurrence of MNOS is consistently observed without tumor association, in contrast to the high prevalence of paraneoplastic NMDARE associated with ovarian teratoma, suggestive of differences in underlying etiological and pathophysiologic mechanisms [18].
MNOS predominantly affects adult women, with rare pediatric cases reported [21], one study reported four children with episodes of anti-NMDARE and a demyelinating syndrome. Two of them were positive for AQP4-IgG and two of them for MOG-IgG. Additionally, four other children with a diagnosis of anti-NMDARE had extensive white matter lesions on brain MRI without clinical evidence of demyelination. These lessons are resolved over time [7]. Another case report presented a 10-year-old girl with optic neuritis and recurrent encephalitis with focal seizures, both anti-MOG and anti-NMDA antibodies were found in cerebrospinal fluid and serum [20]. Finally, a case of a 10-year-old girl who first presented with anti-NMDARE and later developed four episodes of left optic neuritis. MOG and AQP-4 antibodies were negative [22].
The presence of lesions in the spinal cord is extremely rare. We found only one case report of a pediatric patient with anti-NMDARE who presented with spinal cord lesions. This case report concerned a 15-year-old girl with an initial presentation of typical anti-NMDARE, followed by atypical relapses consistent with seronegative NMSOD [23], A summary of pediatric patients is shown in Table 1.
|
Source |
Age of onset (years) |
Sex |
Features of anti-NMDARE |
Features of demyelination pathology |
MRI |
Interval (months) |
||
|
|
|
|
Clinical |
Anti-NMAR-Antibodies (CSF/serum) |
Clinical |
Other antibodies (CSF/serum) |
|
|
|
Case presented in this article |
13 |
F |
Seizures, memory deficit, apathy and automatisms |
+/- After relapse: +/+ |
/ |
AQP4 -/- MOG -/- |
Multiple supratentorial FLAIR/T2 hyperintense white matter lesions and T2 hyperintense intramedullary lesions at levels C2 and T11 |
8 |
|
Titulaer M et al. 2014 [7] |
8 |
F |
Fluctuating consciousness, seizures, dystonia and orofacial dyskinesias |
+/+ |
1st: bilateral ON
2nd: LETM |
AQP4 +/+ MOG n.d./- |
Multifocal increased T2/FLAIR signal in putamen, internal capsulae, subcortical insula, hippocampi and temporal regions |
84 |
|
|
13 |
F |
Seizures, altered behavior, memory and speech dysfunction |
+/n.d. |
Recurrent LETM |
AQP4 +/n.d. MOG -/n.d. |
Transient mild FLAIR increased signal, from medulla to C5 |
11 |
|
|
17 |
M |
Behavioral dysfunction, insomnia, mutism, catatonia, dyskinesias |
+/n.d. |
1st: facial palsy, ataxia
2nd: ataxia, ophtalmo-plegia |
AQP4 -/n.d.
MOG +/n.d. |
T2/FLAIR increased signal in right frontal/temporal lobes and left parietal/temporal region |
3 |
|
|
10 |
M |
Seizures, fever |
+/- |
Hemiparesis, bilateral visual impairment |
AQP4 n.d./- MOG +/+ |
Increased T2/FLAIR signal in the grey matter of frontal lobes, multiple areas of increased T2/FLAIR signal in white matter |
96 |
|
|
16 |
F |
Headache, diplopia, anisocoria, vertigo, ataxia, tremor, memory deficit, dyskinesia, hemiparesis |
+/- |
/ |
AQP4 -/+ MOG -/- |
Multiple increased T2/FLAIR signals in the right mesotemporal lobe, cerebellum and thalamus |
/ |
|
|
4 |
F |
Seizures, hemiparesis, mutism, chorea, orofacial dyskinesia |
+/n.d. |
/ |
AQP4 n.d./- MOG +/+ |
Multifocal areas of T2/FLAIR increased signal |
/ |
|
|
6 |
M |
Personality change, hypersomnia |
+/- |
/ |
AQP4 n.d./- MOG +/+ |
Multifocal areas of increased FLAIR signal: cortex, subcortex, thalamus, basal ganglia, cerebellum, brainstem, cervical/thoracic cord |
/ |
|
|
13 |
F |
Confusion, behavioral dysfunction, stupor, orofacial dyskinesia |
+/+ |
/ |
AQP4 n.d./- MOG -/- |
Multifocal subcortical and periventricular T2/FLAIR increased signal abnormalities |
/ |
|
Taraschenko O et al. 2019 [20] |
10 |
F |
Seizures |
+/- |
Started with bilateral ON |
AQP4 -/- MOG -/+ |
Right parietal cortical hyperintensity on FLAIR sequences consistent with cortical edema |
13 |
|
Kruer M et al. 2010 [23] |
15 |
F |
Headache, seizures, encephalopathy, orofacial dyskinesia |
+/- |
Recurrent episodes of LETM and ON |
AQP4 -/- MOG -/- |
Multiple contrast enhancing lesions (brain/spine) |
1 |
|
Ishikawa N et al. 2007 [22] |
10 |
F |
Seizures |
+/+ |
Recurrent ON |
-/- -/- |
Gray matter with white matter lesions in the frontal lobe on FLAIR and gadolinium-enhancing white matter lesions in the parietal lobe and in the left optic nerve on T 1 |
/ |
|
M: Male; F: Female; n.d.: not done |
||||||||
There are a few additional cases reported in adults, including four young women with anti-NMDARE associated with ovarian teratoma, of whom only one exhibited T2 hyperintensity in the dorsal aspect of the medulla and three similar areas in the spinal cord [24]. It is crucial to continue monitoring for ovarian teratomas since they may become detectable even after a period of up to three years following the diagnosis of anti-NMDARE.
Others reported a case involving a 31-year-old woman who experienced sensory symptoms in her feet, which gradually progressed to her legs and lower abdomen. MRI of the spinal cord revealed focal and segmental areas of high T2-signal intensity. Besides her sensory symptoms, which were attributed to the myelitis seen on MRI and resolved spontaneously, she developed significant neuropsychiatric symptoms and seizures consistent with anti-NMDARE, confirmed by positive antibodies in both serum and cerebrospinal fluid. After treatment with high-dose corticosteroids, repeat imaging showed partial resolution of these lesions [25].
For our patient, we followed the treatment algorithm from the "International Consensus Recommendations for the Treatment of Paediatric NMDAR Antibody Encephalitis” [26]. Treatment options vary based on the severity of the case. Our patient was classified as "severe" due to her inability to self-care, communicate adequately, and her general unresponsiveness to her immediate environment. Consequently, we rapidly escalated to second-line immunotherapy, which resulted in good clinical outcomes in the short term. Relapse was seen after 8 months, with again good result after rituximab administration. The choice for rituximab as preferred second-line immunotherapy in possible overlap syndromes still needs to be investigated but makes sense considering the role of B-cells in antibody-mediated disorders and the overwhelming evidence for B-cell monoclonal antibodies in the treatment of multiple sclerosis.
Conclusion
Concurrent NMDARE and MS are rare but increasingly recognized and may provide a unique insight into common pathophysiological mechanisms in two seemingly different autoimmune disorders. Treatment with rituximab may be preferable in these overlap syndromes, although long-term data are needed to provide insights in prognosis. It is clear that a long-term follow-up investigation is necessary due to the prolonged interval between the onset of NMDARE and the development of demyelinating diseases.
Conflicts of Interest
The authors have no conflicts of interest to disclose.
Funding Statement
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
2. Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008 Dec;7(12):1091-8.
3. Guasp M, Dalmau J. Encephalitis associated with antibodies against the NMDA receptor. Med Clin (Barc). 2018 Jul 23;151(2):71-9.
4. Stawicka E. Anti-NMDA receptor encephalitis - the narrative review of literature with particular regard to pediatric population. Psychiatr Pol. 2022 Dec 31;56(6):1315-26.
5. Yuan L, Mao G, Zhang Y, Xu Y, Chen Q, Shan B, et al. Typical metabolic pattern of 18F-FDG PET in Anti-NMDAR encephalitis in the acute and subacute phases and its correlation with T2 FLAIR-MRI features. BMC Neurosci. 2023 Sep 25;24(1):51.
6. Jan S, Anilkumar AC. Atypical Brain MRI Findings in a Child with Delayed Diagnosis of Anti-N-Methyl-D-Aspartate Receptor Encephalitis. Cureus. 2021 Sep 19;13(9): e18103.
7. Titulaer MJ, Höftberger R, Iizuka T, Leypoldt F, McCracken L, Cellucci T, et al. Overlapping demyelinating syndromes and anti–N-methyl-D-aspartate receptor encephalitis. Ann Neurol. 2014 Mar;75(3):411-28.
8. Chen LW, Olivé-Cirera G, Fonseca EG, Mistieri Simabukuro M, Iizuka T, Armangue T, et al. Very Long-Term Functional Outcomes and Dependency in Children with Anti-NMDA Receptor Encephalitis. Neurol Neuroimmunol Neuroinflamm. 2024 May;11(3): e200235.
9. Wang X, Yin Y, Wang X, Xu G, Tian J, Ma X. White matter microstructural alterations in patients with anti-N-methyl-D-aspartate receptor encephalitis: A tract-based spatial statistics study. Mult Scler Relat Disord. 2024 Apr; 84:105500.
10. Phillips OR, Joshi SH, Narr KL, Shattuck DW, Singh M, Di Paola M. Superficial white matter damage in anti-NMDA receptor encephalitis. J Neurol Neurosurg Psychiatry. 2018 May;89(5):518-25.
11. Bartels F, Krohn S, Nikolaus M, Johannsen J, Wickström R, Schimmel M, et al. Clinical and Magnetic Resonance Imaging Outcome Predictors in Pediatric Anti-N-Methyl-D-Aspartate Receptor Encephalitis. Ann Neurol. 2020 Jul;88(1):148-59.
12. Lebrun-Frenay C, Kantarci O, Siva A, Sormani MP, Pelletier D, Okuda DT; 10-year RISC study group on behalf of SFSEP, OFSEP. Radiologically Isolated Syndrome: 10-Year Risk Estimate of a Clinical Event. Ann Neurol. 2020 Aug;88(2):407-17.
13. Takeda A, Shimada H, Tamura A, Yasui M, Yamamoto K, Itoh K, et al. A case of anti-N-methyl-d-aspartate receptor encephalitis with multiple sclerosis-like demyelinated lesions. Mult Scler Relat Disord. 2014 May;3(3):391-7.
14. Liu P, Yan H, Li H, Zhang C, Li Y. Overlapping anti-NMDAR encephalitis and multiple sclerosis: A case report and literature review. Front Immunol. 2023 Jan 30; 14:1088801.
15. Zhou R, Jiang F, Cai H, Zeng Q, Yang H. Case Report: Antibodies to the N-Methyl-D-Aspartate Receptor in a Patient with Multiple Sclerosis. Front Immunol. 2021 Apr 23; 12:664364.
16. Yang S, Wu Y, Sun L, Ma M, Ou S, Meng Y, et al. White matter abnormalities and multivariate pattern analysis in anti-NMDA receptor encephalitis. Front Psychiatry. 2022 Sep 23; 13:997758.
17. Kuchling J, Penner L, Ceballos RA, Maier L, Tietz A, Rapp D, et al. NMDA Receptor Encephalitis and Multiple Sclerosis Overlap Syndrome-Part I: Clinical Findings and MRI Characteristics.
18. Chen W, Li Q, Wang T, Fan L, Gao L, Huang Z, et al. Overlapping syndrome of anti-N-methyl-D-aspartate receptor encephalitis and anti-myelin oligodendrocyte glycoprotein inflammatory demyelinating diseases: A distinct clinical entity? Mult Scler Relat Disord. 2021 Jul; 52:103020.
19. Zoccarato M, Saddi MV, Serra G, Pelizza MF, Rosellini I, Peddone L, et al. Aquaporin-4 antibody neuromyelitis optica following anti-NMDA receptor encephalitis. J Neurol. 2013 Dec;260(12):3185-7.
20. Taraschenko O, Zabad R. Overlapping demyelinating syndrome and anti-N-methyl-d-aspartate receptor encephalitis with seizures. Epilepsy Behav Rep. 2019 Oct 25; 12:100338.
21. Ran Y, Wang L, Zhang F, Ao R, Dong Z, Yu S. Anti-NMDAR encephalitis followed by seropositive neuromyelitis optica spectrum disorder: A case report and literature review. Clin Neurol Neurosurg. 2017 Apr; 155:75-82.
22. Ishikawa N, Tajima G, Hyodo S, Takahashi Y, Kobayashi M. Detection of autoantibodies against NMDA-type glutamate receptor in a patient with recurrent optic neuritis and transient cerebral lesions. Neuropediatrics. 2007 Oct;38(5):257-60.
23. Kruer MC, Koch TK, Bourdette DN, Chabas D, Waubant E, Mueller S, et al. NMDA receptor encephalitis mimicking seronegative neuromyelitis optica. Neurology. 2010 May 4;74(18):1473-5.
24. Vitaliani R, Mason W, Ances B, Zwerdling T, Jiang Z, Dalmau J. Paraneoplastic encephalitis, psychiatric symptoms, and hypoventilation in ovarian teratoma. Ann Neurol. 2005 Oct;58(4):594-604.
25. Pennington C, Livingstone S, Santosh C, Razvi S. N-methyl D-aspartate receptor antibody encephalitis associated with myelitis. J Neurol Sci. 2012 Jun 15;317(1-2):151-3.
26. Nosadini M, Thomas T, Eyre M, Anlar B, Armangue T, Benseler SM, et al. International Consensus Recommendations for the Treatment of Pediatric NMDAR Antibody Encephalitis. Neurol Neuroimmunol Neuroinflamm. 2021 Jul 22;8(5): e1052.