Abstract
Cytokine release syndrome represents a significant barrier to the widespread application of chimeric antigen receptor (CAR)-T cell therapies. We performed a broad analysis of preclinical and clinical studies that tested different designs of the CAR construct to tune CAR signaling, with an emphasis on effects of CAR designs on cytokine release from activated CAR-T cells. Evidence from these studies has shown that CAR signal strength and induced cytokine release can be effectively tuned by choosing different antigenbinding domains, hinge and transmembrane regions, costimulatory domains, and activation domains of a CAR construct. The detailed understanding in this aspect will pave the way to develop CAR-T cell products that exert robust anti-cancer function without the exceeding release of cytokines, thus fulfilling their promise in cancer therapy.
Introduction
Chimeric antigen receptor (CAR)-T cell therapy is nowadays at the forefront of cancer treatment. The discovery of CAR could be traced back to Gross’s study in 1989, who firstly transfected T cells by using an integrated T-cell receptor’s (TCR) constant domain with the antibody’s variable domain and showed the specific cytotoxic effects of these cells towards the antigen-bearing target cells [1]. Hitherto CAR has been further developed, with typically the construct of an extracellular antigen binding domain, a hinge domain, a transmembrane domain, a co-stimulatory domain, an activation domain, and the other relevant stimulation molecules [2]. In the clinical setting, there are four CD19 and one BCMAdirected CAR-T therapies approved by US FDA in treating large B cell lymphoma, mantle cell lymphoma, and multiple myeloma [3,4], demonstrating a significant development of CAR-T treatments. Meanwhile, numerous clinical trials are currently ongoing to evaluate the safety and efficiency of other CAR-T treatments in both leukemia and solid tumors [5], indicating the huge potential of the cell therapy approach in malignancy control.
Although the effectiveness of CAR-T cell therapy in blood cancer treatment has been proven, the related toxicities and safety concerns were also observed in patients and may restrict the widespread use of CAR-T cell therapies. One of the most prevalent side effects is known as the cytokine release syndrome (CRS). The clinical presentation of CRS could be very diversified, from the mild symptoms such as fever and arthralgia, to the much more severe cases with multi-organ system failure or even death [6]. CRS is typically caused by massive in vivo CAR-T cell activation and the associated excessive secretion of the inflammatory cytokines such as interleukin (IL)-6, IL-2, interferon (IFN)-γ, tumor necrosis factor (TNF)-α by CAR-T cells or other activated immune cells during the treatment [7,8]. However, some of these cytokines are also important in the process of CAR-T activation and can potentiate the effects of CAR-T cell therapy against malignancy. Therefore, finetuning CAR-T cell signaling to balance cytokine secretion is vital to establish a CAR-T cell therapeutic window that exerts powerful anti-cancer function without the exceeding release of the cytokines.
Evidence from the currently published studies clearly indicates that CAR signal strength can be manipulated by all the components of a CAR structure, from choosing CAR antigen recognition and binding domains with different affinity, designing CAR hinge and transmembrane regions capable of modulating different levels of cytokine secretion, selecting costimulatory domains that alter cytokine production and CAR-T cell persistence, and engineering the immunoreceptor tyrosine-based activation motif (ITAM)s in the activation domains [9,10] (Figure 1). While the severity of CRS is certainly affected by multiple factors such as the tumor burden and administered doses [6], the amendment of different CAR structural components appears to be another way to ameliorate cytokineassociated toxicities of CAR-T cell therapy. Here, we summarize the related studies and discuss the implications of the findings for future CAR-T cell therapies.
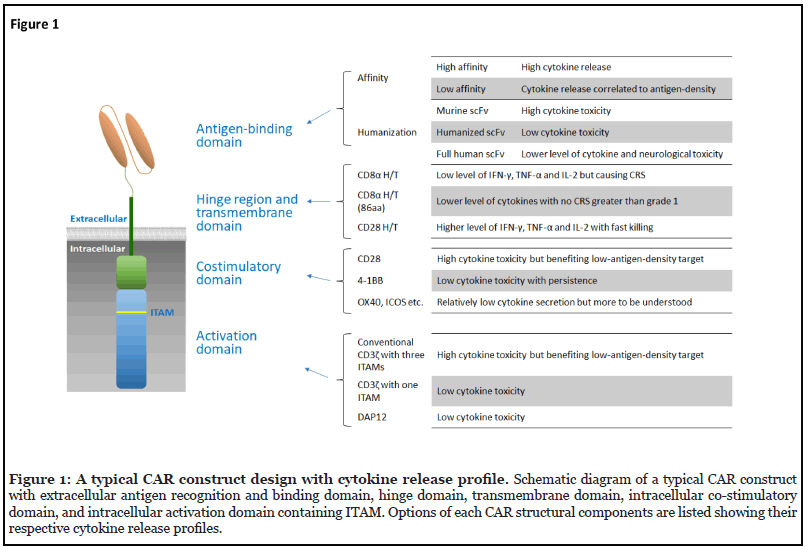
Antigen-Binding Domain
The antigen recognition and binding domain is generally designed to recognize and bind the tumor-associated or -specific surface antigens on cancer cells, triggering a subsequent major histocompatibility complex (MHC) independent T cell activation. This domain is usually adapted from a monoclonal antibody’s heavy and light chains, named single chain variable fragments (scFvs). Besides, some ectodomains of native immune cell receptors could also be directly used as an antigen-binding domain, such as the NKG2D receptor on natural killer (NK) cells.
Other than sensing the tumor antigens, the affinity of antigen-binding domain has been unveiled to be associated with the cytokine related toxicities. Liu et al. found that the affinity of the scFv region could be favorably used to regulate the secretion of the inflammatory cytokines [11]. In the case of targeting ErbB2 antigen-expressing cell lines, CAR-T cells with a high affinity antigen-binding domain was reactive against both high and low levels of ErbB2 expression, but secreted even greater levels of IFN-γ and IL-2 when targeting a low level of ErbB2 expression. In contrast, CAR-T cells with a low affinity antigen-binding domain were interestingly more sensitive to ErbB2 overexpressed cancer cells with more cytokine secretion, indicating its superior specificity to ErbB2 overexpressing tumor cells but not the normal somatic cells with a physiologic level of ErbB2 expression [11]. This finding is consistent with another study using anti-EGFR CAR-T cells, which demonstrated a reduction of IFN-γ releasing with the low affinity anti-EGFR CAR-T cells targeting the low-density EGFR cancer cells (to mimic the normal human cells) compared to the high affinity anti-EGFR CAR-T cells in vivo [12]. Meanwhile, both high and low affinity anti-EGFR CAR-T cells effectively controlled the growth of high-intensity EGFR+ glioma, but the treatment with low affinity anti-EGFR CAR-T cells conferred to no apparent toxicity and extended the mice survival for almost 80% [12].
Moreover, scFv humanization in antigen-binding domain has been shown to play a crucial role in regulating the cytokine release of CAR-T cells. Song et al. showed that the replacement of the murine Mov-19 scFv-based folate receptor-alpha (αFR) CAR to a fully human anti-αFR C4 CAR could retain the anti-tumor activity in vitro and in vivo but reduce the off-tumor toxicity with a less IFN-γ secretion [13]. In a recent phase I clinical trial, Kochenderfer’s lab at National Institutes of Health (NIH) demonstrated an anti-B cell lymphoma activity of a new type of anti-CD19 CAR-T cells with fully human binding domains similar to that of anti-CD19 CAR-T cells containing murine binding domains, but a significantly lower level of neurological toxicity [14]: only 5% when the former were used vs. 50% in the latter’s case. The findings that the anti-CD19 CAR-T cells with fully human binding domains released lower serum levels of cytokines than the anti-CD19 CAR-T cells containing murine binding domains in patient blood samples explain the improved safety profile [14]. This clinical study has clearly shown that the CD19-speficific CAR therapy is not inherently associated with the cytokine toxicity and that the incorporation of fully human binding domains in CARs is crucial in mitigating the toxicity. When a fully human scFv is not available, a recently published study by Dwivedi et al. demonstrated that humanizing the framework region of scFv derived from a murine anti- CD19 monoclonal antibody was also effective in reducing cytokine release, even inducing less IL-6 secretion from monocytes, while maintaining the anti-tumor activity [15]. Such a cytokine release profile would benefit CAR-T therapy with a low risk of CRS.
Above studies explored the manipulation of the CAR antigen-binding domain and provided clues for adopting scFvs with low affinity, fully human sequence, or humanized sequence in a safe CAR design. Such designs should be the first consideration to develop a new CAR with a low risk of the cytokine-mediated toxicity.
Hinge Region and Transmembrane Domain
The hinge region and the transmembrane (H/T) domains are the linker between the extracellular antigen binding domain and the intracellular signaling transduction domain in the CAR construct. The hinge region supports stable CAR expression and provides spatial flexibility in binding to target antigens, while the transmembrane domain is mainly responsible for anchoring the CAR construct on the cell surface [16]. These two regions are usually obtained directly from a portion of widely expressed receptors on T cells, such as CD8 and CD28, and are likely to be the least understood for their regulatory roles in controlling cytokine release. However, a few papers recently reported the selection of H/T domains may not only affect the CAR expression and signaling intensity, but also lead to different degrees of cytokine release [14,17-20].
Also, from Kochenderfer’s lab at NIH, Alabanza et al. were the first to demonstrate that manipulation of H/T domains can mitigate the side effect of massive cytokine release from anti-CD19 CAR-T cells containing an scFv with fully human variable regions [18]. By utilizing the CD8α H/T domains instead of CD28 H/T domains, much lower level of IFN-γ, TNF-α and IL-2 were produced by the stimulated anti-CD19 CAR-T cells, while the degranulation, cytotoxicity and proliferation were kept at similar levels [18]. The clinical advantage of this CAR construct has been confirmed in a phase I dose-escalation trial [14], in which the anti-CD19 CAR-T cell product with the CD8α H/T region caused a 5% incidence rate of Grade 3 or 4 neurologic toxicity, strikingly different from 55% of Grade 3 or 4 neurologic toxicity observed in a previous clinical trial using anti-CD19 CAR-T cells that incorporated CD28 H/T domains.
In another phase I clinical trial, Ying et al. reported that slight modification of the amino acid sequence of a H/T region was enough to improve the safety profiles of an anti-CD19 CAR-T cell therapy [19]. Based on the FDA approved prototype CTL019 (Kymriah) CAR construct (CD19-BBz(71)), they generated a new anti-CD19 CAR molecule (CD19-BBz(86)) by changing the sequence encoding the CD8α H/T domain from the original 71 amino acids to 86 amino acids. This modification by adding 15 amino acid residues from the native CD8α molecule to the CD8α H/T domain altered the predicted structure of the CAR and resulted in no change in cytolytic activity but less cytokine secretion and reduced CAR-T proliferation in vitro [19]. Similarly, while being effective in eliminating CD19-positive tumor burden in a mouse model, the CD19-BBz(86) CAR-T cells did not cause CRS and all mice survived healthily through the in vivo experiment. On the other hand, the CD19-BBz(71) CAR-T cells caused severe CRS with significant weight lost in a large portion of treated mice and eventually animal deaths [19]. In patients with B cell lymphoma, the CD19-BBz(86) CAR-T cell therapy provided 54.5% complete remission and 18% partial remission when the highest dose of 2-4 × 108 cells was used [19]. More importantly, no significant increment of serum cytokine level including IL-6, TNF-α, IFN-γ, IL-17A, IL-2, IL-15, IL-5, IL-12 p70, IL-1β and C-reactive protein (CRP), no CRS greater than Grade 1 and no neurological toxicities were observed in this trial [19]. Thus, altering non-signaling H/T domains can potentially reduce the incidence of serious cytokine release-associated toxicities in patients.
In a further twist to the effects of non-signaling H/T domains on CAR-T cell activity, Majzner et al. recently showed that tuning of H/T domains could enhance the sensitivity of CAR-T cells against low level of antigen expression to prevent tumor escape [20]. In their study, anti-CD19 CAR-T cells incorporated with CD28 H/T domains was more reactive to low-antigen-density tumors in vitro and in vivo, compared to a counterpart setting with a CD8 hinge and transmembrane domain, producing higher level of IL-2 and performing fast killing [20]. The superior anti-tumor functions of CAR-T cells with a CD28 H/T region as compared with a CD8 H/T region were further observed in two solid tumor models used to test CAR-T cells against HER2-positive breast cancer and B7- H3 (CD276)-positive neuroblastoma [20]. They revealed that the CD28 H/T domain could promote the receptor and signaling molecules clustering with better spatial organization but a more moderate activation kinetics, which is expected to benefit the cytokine release profile.
Taken together, harnessing the effects of H/T domains on the CAR-T cell activation related cytokine release could be an attractive way to improve the efficacy and safety profile of a CAR construct without touching its antigenbinding domain. This could be important in developing a CAR against solid tumors where the expression of tumorassociated antigens is highly variable, thus the specificity and affinity of an antigen-binding domain could be crucial to limit on-target-off-tumor toxicities.
Costimulatory Domain
In natural T cells, several costimulatory molecules, such as CD28 and 4-1BB (CD137), express on the cell surface to assist the signal transduction of the T cell receptor (TCR). The endodomains of these naturally presented costimulatory molecules have been used in the 2nd and 3rd generation CAR constructs to improve functions, given the findings that the TCR activation domain CD3ζ alone in the first generation CAR construct was not sufficient to sustain the CAR-T cell response and proliferation in vivo [21]. Currently, the CD28 and 4-1BB endodomains are the most well-established CAR costimulatory domains, with the five FDA approved CAR-T therapies using either one of them [22-26].
Typically, CAR-T cells equipped with CD28 or 4-1BB resulted in distinct cytokine release phenotypes. As shown in Salter et al.’s study, CD28 CAR-T cells usually conferred to short term effector responses and secreted markedly higher inflammatory cytokines such as TNF-α compared to the 4-1BB CAR-T cells, which may lead to cytokine toxicities [27]. In the mouse model, CD28 CAR-T cells were demonstrated to be less capable in eliminating Raji lymphoma cells and more easily to enter an exhausted phenotype [27]. In clinical trials with anti-CD19 CAR-T cells, CRS occurrence was 93% in the CD28 CAR-T treatment, but 57% in 4-1BB CAR-T treatment [6,28-30]. These findings all together suggest that 4-1BB CAR-T might be safer compared to CD28 CAR-T in consideration of cytokine-related toxicity.
However, assisted with the production of a higher amount of cytokines, CAR-T cells with CD28 costimulatory domains displayed superior anti-tumor activity against low-antigen-density targets comparing to CAR-T cells equipped with 4-1BB costimulatory domains, whereas CAR-T cells with 4-1BB costimulatory domains displayed attenuated response to low-antigen-density tumors due to insufficient CAR signaling [20]. Nevertheless, the 4-1BB CAR signaling can be augmented against low-antigendensity tumors while maintaining its unique capacity for CAR-T cell persistence, for example by adding additional ITAMs as discussed below [20].
The endodomains of many other co-stimulatory molecules, such as OX40, ICOS (CD278) and CD27, have also been tested in CAR constructs as a costimulatory domain but they are mostly not thoroughly investigated yet to solve the paradox between the effectiveness and safety concerns. One study showed that GD2-specific CAR-T cells with the OX40 costimulatory domain led to less IFN-γ and IL-2 secretion, but the antitumor activities were also hindered in vivo [31]. Moreover, the ICOS costimulatory domain in a mesothelin-specific CAR was shown to confer a similar secretion level of IFN-γ but lower levels of IL-2 and TNF-α upon the CAR-T cell activation as compared to the CD28 costimulatory domain, whereby the CAR-T cells with 4-1BB domain secreted all three cytokines in the lowest level [32].
In short, the current understanding of the costimulatory domain function in CAR-T cells against liquid and solid tumors supports the notion that the choice of a costimulatory domain has an enormous effect on not only CAR signal strength and CAR-T cell persistence, but also the cytokine release profile of CAR-T cells.
ITAMs in a CAR Activation Domain
The activation domain of a CAR is the primary signaling domain to trigger the intracellular cascade signaling and lead to subsequent anti-tumor effects. The most commonly used activation domain in the CAR construct is the cytoplasmic tail of TCR CD3ζ, containing three immunoreceptor tyrosine-based activation motifs (ITAMs), which can be phosphorylated to recruit Syk kinase and activate downstream signaling pathways [33].
A recent paper by Feucht et al. has demonstrated that the number of the ITAMs in the CD3ζ domain can be manipulated to optimize CAR-T functions. They had shown that the anti-CD19 CAR-T cells with only one functional ITAM was able to induce rapid tumor eradication in vivo and outperformed the triple or double ITAM-containing CAR-T cells [34]. The single ITAM configuration of the anti- CD19 CAR-T cells supported the CAR-T cell persistence by balancing the replicative capacity of long-lived memory cells and the acquisition of effective antitumor function. Moreover, the reduction in cytokine release was also observed from two out of three single ITAM configurations tested in their CD3ζ CAR-T cells [34].
The detailed studies performed by Majzner et al. further demonstrated the importance of the number of ITAMs in a CD3ζ domain in regulating CAR-T cell function [20]. They developed an anti-CD19 4-1BB CAR that incorporated two copies of CD3ζ, “double zeta” with the total six ITAMs. When the functionality of “double zeta” versus “single zeta” CAR-T cells against target cells with lower antigen densities were evaluated, more IL-2 molecules were generated in response to antigen stimulation in “double zeta” versus “single zeta” CARs, suggesting that including additional ITAMs in the CAR can enhance signal strength. They further observed that anti-CD19 4-1BB CAR-T cells expressing only one CD3ζ ITAM (the most membraneproximal) displayed the reduced ability to produce IL-2 in response to tumor cells expressing low levels of CD19 compared with identical CARs with intact three ITAMs [20]. These findings emphasize the crucial roles of the number of ITAMs in a CAR in cytokine release and finetuning CAR-T cell signaling.
Other than the modification of the CD3ζ activation domain, we designed a de novo single chain CAR construct by fusing the NKG2D receptor ectodomain with a 4-1BB costimulatory domain and a DAP12 endodomain [35]. DAP12 is a short transmembrane protein containing a single ITAM only, the phosphorylation of which results in the recruitment of the cytoplasmic ZAP70 tyrosine kinases and downstream signaling [36]. The co-expression of the full-length DAP12 molecule with other signaling domains of a CAR construct was confirmed previously to be feasible and effective in improving the antitumor activity of T cells [37,38]. Our study focused on the DAP12 endodomain, especially on the effects of the single ITAM on cytokine release. We generated and compared three different CAR constructs: NKG2Dbp CAR containing the DAP12-drived activation domain with a single ITAM per CAR molecule, NKG2Dbz CAR containing the CD3ζ- drived activation domain with three ITAMs per CAR molecule, and NKG2Dbz1 CAR containing the first ITAM of the CD3ζ chain only (the most membrane-proximal), an ITAM with a sequence different from the one in DAP12. We demonstrated the significant lower levels of in vitro secretion of IL-2, IFN-γ and TNF-α from the NKG2Dbp CAR-T cells as compared to the activated NKG2Dbz CAR-T and NKG2Dbz1 CAR-T cells without decreasing in vitro tumor cell lytic activity. In a mouse model with established human colorectal xenografts, both NKG2Dbp CAR-T and NKG2Dbz CAR-T cells exhibited similar tumor eradicating efficacies: with one single dose of CAR-T injection, most of the mice became cancer-free. However, body weight lost (>15%) and xenogeneic graft-versus-host disease (x-GVHD) were observed in the NKG2Dbz CAR-T treated mice but not in NKG2Dbp CAR-T treated mice. Meanwhile, higher serum concentrations of IL-2, TNF-α, and IFN-γ and x-GVHD associated hepatic coagulative necrosis were detected only in the NKG2Dbz treated mice but not in the NKG2Dbp group [35]. Our findings demonstrated again that cytokine release upon CAR activation can be controlled by decreasing the ITAM density per CAR construct. The difference between NKG2Dbp and NKG2Dbz1 further indicates the advantage of the DAP12 ITAM sequence in balancing CAR-T cell signaling to reduce cytokine release yet maintaining anti-tumor activity.
Overall, apart from using conventional CD3ζ in generating CAR-T cells, manipulation of ITAM numbers in a CD3ζ-cotaining CAR or using other T cell activation domains such as DAP12 could be considered in developing new CAR constructs. In this aspect, the effectiveness of the above DAP12-constaining CAR-T cells in controlling solid tumor progression and x-GVHD in an animal model suggests a potential clinical advantage of the DAP12 activation domain in CAR-T cell therapy against solid tumors without inducing excess serum levels of cytokines.
Perspective on CAR Construct Design in Therapy against Solid Tumors
Our understanding on the effects of different CAR structural components in CAR-T cells against solid tumors is just beginning to emerge. More data are essential to developing our insights in this area where it is challenging for CAR-T cell therapy to deal with the restricted tumor trafficking/infiltration, target antigen heterogeneity, and immunologically suppressive tumor microenvironment (TME) of solid tumors. The considerable CAR designs beyond the conventional structures will be necessary to address these barriers [2].
When anti-CD19 CAR-T cells are injected to treat blood cancers, the injected effector cells can quickly access disseminated cancer cells, as well as CD19-positve normal cells, in the circulation, and are extensively activated and readily expanded upon stimulation by the target cells. CAR-T cell therapy against solid tumors faces tough challenges in achieving significant in vivo expansion and long persistence of the injected effector cells, since solid tumor malignancies are regionally localized at specific tissue sites. As such, probably only a few infused CAR-T cells can reach the tumor site and interact with tumor antigen-expressing solid malignant cells, thus limiting the activation and proliferation of CAR-T cells. Under such a situation, some of the released cytokines from the activated CAR-T cells could play important roles in providing local stimulation to CAR-T cell expansion to achieve therapeutic effects. Hence, a profound knowledge of cytokine biology under such a situation is required to exploit the anti-tumor activity of CAR-T cells against solid tumors while keeping cytokine-associated toxicity to a minimum.
Most of the studies discussed above were focused on anti-CD19 CAR-T therapy against B-cell lymphoma and leukemia. Since the physiological expression of CD19 antigen does not represent a major concern of CAR-T safety, the CAR signaling strength-related therapeutic window can be broadened to a level for the CAR to recognize a very low antigen density, thus increasing the sensitivity of the CAR-T therapy to prevent antigen escape [20]. However, on the flip side, the increased efficacy may come with a liability of increased cytokine-associated toxicities, as already observed clinically. In terms of CAR-T cell therapy against solid tumors, since normal tissues would share a physiological level expression of many tumor-associated antigens, the CAR-T therapeutic window has to be stricter to only target antigen-overexpressing tumor cells. This point should be taken into consideration when designing a CAR construct with the balanced cytokine release and anti-cancer activity.
The TME comprises numerous suppressive immune cells, such as regulatory T (Treg) cells, myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages, which release cytokines to promote tumorigenesis and progression. Some of the cytokines deliver T cell inhibitory signals to inhibit CAR-T cell function. Various cytokines can also be released from infiltrating CAR-T cells after activated in the TME by tumor cells. Depending on context and elements in the TME some of them may exert pro- or anti-tumor activities, like double-edged swords [39], making it a formidable challenge to CAR engineering to fine-tune the secretion stimulatory cytokines in the TME.
Unlike the great successes in CAR-T cell therapy against blood cancer, no equivalent successes have been achieved yet in patients with solid tumors. Addressing multiple challenges of the solid tumor TME is an area of active investigation. Augmenting CAR-T cells by altering CAR construct design to balance cytokine secretion could be a possible way to explore.
Concluding Remarks
The above discussed reports convincingly demonstrate the interplays between different components of the synthetic CAR construct and their effects on T cell function. Obviously, all of the structural components are crucial in CAR design and even minor changes in some of these structures can tune the signaling strength required for optimal CAR-T cell activity. Our discussion concentrated on how to manipulate the components to tune cytokine release from the activated CAR-T cells, thus potentially mitigating the risk of CRS. However, tuning cytokine release would more or less affect CAR signal intensity and may further affect the efficacy of CAR-T cells, especially when facing the impediments posed by the TME of solid tumors. Nevertheless, precise engineering of the CAR constructs to achieve a striking delicate balance between anti-tumor efficacy and controlled cytokine-mediated toxicity is certainly required for providing effective yet safe cancer treatment.
Acknowledgement
This work was supported by the Singapore Ministry of Health’s National Medical Research Council (NMRC/ OFLCG/003/2018; MOH-000465-01) and Agency for Science, Technology and Research, Singapore (IAFPP: H19/01/a0/022).
Authors’ Contributions
Conceptualization, W.S.; investigation, D.Z. and Z.S.; resources, N.Y.Y.; writing—original draft preparation, D.Z.; writing—review and editing, Z.S. and W.S.; supervision and funding acquisition, W.S. All authors have read and approved the final manuscript.
References
2. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. 2020;17(3):147-67.
3. Mullard A. FDA approves fourth CAR-T cell therapy. Nat Rev Drug Discov. 2021;20(3):166.
4. Mullard A. FDA approves first BCMA-targeted CAR-T cell therapy. Nat Rev Drug Discov. 2021.
5. Holzinger A, Abken H. Advances and Challenges of CAR T Cells in Clinical Trials. Recent Results Cancer Res. 2020;214:93-128.
6. Shimabukuro-Vornhagen A, Godel P, Subklewe M, Stemmler HJ, Schlosser HA, Schlaak M, et al. Cytokine release syndrome. J Immunother Cancer. 2018;6(1):56.
7. Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016;6(6):664-79.
8. Brudno JN, Kochenderfer JN. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019;34:45-55.
9. Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer. 2021;21(3):145-61.
10. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11(4):69.
11. Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015;75(17):3596- 607.
12. Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res. 2015;75(17):3505-18.
13. Song DG, Ye Q, Poussin M, Liu L, Figini M, Powell DJ, Jr. A fully human chimeric antigen receptor with potent activity against cancer cells but reduced risk for off-tumor toxicity. Oncotarget. 2015;6(25):21533-46.
14. Brudno JN, Lam N, Vanasse D, Shen YW, Rose JJ, Rossi J, et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat Med. 2020;26(2):270-80.
15. Dwivedi A, Karulkar A, Ghosh S, Srinivasan S, Kumbhar BV, Jaiswal AK, et al. Robust anti-tumor activity and low cytokine production by novel humanized anti- CD19 CAR-T cells. Mol Cancer Ther. 2021.
16. Guedan S, Calderon H, Posey AD, Jr., Maus MV. Engineering and Design of Chimeric Antigen Receptors. Mol Ther Methods Clin Dev. 2019;12:145-56.
17. Fujiwara K, Tsunei A, Kusabuka H, Ogaki E, Tachibana M, Okada N. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells. 2020;9(5).
18. Alabanza L, Pegues M, Geldres C, Shi V, Wiltzius JJW, Sievers SA, et al. Function of Novel Anti-CD19 Chimeric Antigen Receptors with Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Mol Ther. 2017;25(11):2452-65.
19. Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, et al. A safe and potent anti-CD19 CAR T cell therapy. Nat Med. 2019;25(6):947-53.
20. Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, et al. Tuning the Antigen Density Requirement for CAR T-cell Activity. Cancer Discov. 2020;10(5):702-23.
21. Brocker T, Karjalainen K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J Exp Med. 1995;181(5):1653-9.
22. Summary Basis for Regulatory Action - YESCARTA: U.S. Food & Drug Administration.; 2017. Available from: https://www.fda.gov/media/108788/download
23. Summary Basis for Regulatory Action - KYMRIAHTM: U.S. Food & Drug Administration.; 2018. Available from: https://www.fda.gov/media/113215/download
24. Summary Basis for Regulatory Action - TECARTUS: U.S. Food & Drug Administration.; 2020. Available from: https://www.fda.gov/media/141093/download
25. Summary Basis for Regulatory Action - BREYANZI: U.S. Food & Drug Administration.; 2021. Available from: https://www.fda.gov/media/146242/download
26. Summary Basis for Regulatory Action - ABECMA: U.S. Food & Drug Administration.; 2021. Available from: https://www.fda.gov/media/147627/download
27. Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A, Alderman EJ, et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci Signal. 2018;11(544).
28. Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell. 2015;28(4):415- 28.
29. Ying Z, He T, Wang X, Zheng W, Lin N, Tu M, et al. Parallel Comparison of 4-1BB or CD28 Co-stimulated CD19-Targeted CAR-T Cells for B Cell Non-Hodgkin's Lymphoma. Mol Ther Oncolytics. 2019;15:60-8.
30. Zhao X, Yang J, Zhang X, Lu XA, Xiong M, Zhang J, et al. Efficacy and Safety of CD28- or 4-1BB-Based CD19 CAR-T Cells in B Cell Acute Lymphoblastic Leukemia. Mol Ther Oncolytics. 2020;18:272-81.
31. Quintarelli C, Orlando D, Boffa I, Guercio M, Polito VA, Petretto A, et al. Choice of costimulatory domains and of cytokines determines CAR T-cell activity in neuroblastoma. Oncoimmunology. 2018;7(6):e1433518.
32. Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood. 2014;124(7):1070-80.
33. Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T cells. Biomark Res. 2017;5:22.
34. Feucht J, Sun J, Eyquem J, Ho YJ, Zhao Z, Leibold J, et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med. 2019;25(1):82-8.
35. Ng YY, Tay JCK, Li Z, Wang J, Zhu J, Wang S. T Cells Expressing NKG2D CAR with a DAP12 Signaling Domain Stimulate Lower Cytokine Production While Effective in Tumor Eradication. Mol Ther. 2021;29(1):75-85.
36. Lanier LL. DAP10- and DAP12-associated receptors in innate immunity. Immunol Rev. 2009;227(1):150-60.
37. Chen B, Zhou M, Zhang H, Wang C, Hu X, Wang B, et al. TREM1/Dap12-based CAR-T cells show potent antitumor activity. Immunotherapy. 2019;11(12):1043-55.
38. Wang E, Wang LC, Tsai CY, Bhoj V, Gershenson Z, Moon E, et al. Generation of Potent T-cell Immunotherapy for Cancer Using DAP12-Based, Multichain, Chimeric Immunoreceptors. Cancer Immunol Res. 2015;3(7):815- 26.
39. Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Perez-Gracia JL, et al. Cytokines in clinical cancer immunotherapy. Br J Cancer. 2019;120(1):6-15.