Keywords
COVID-19, Brain, neurological, immunity, bystander
Commentary
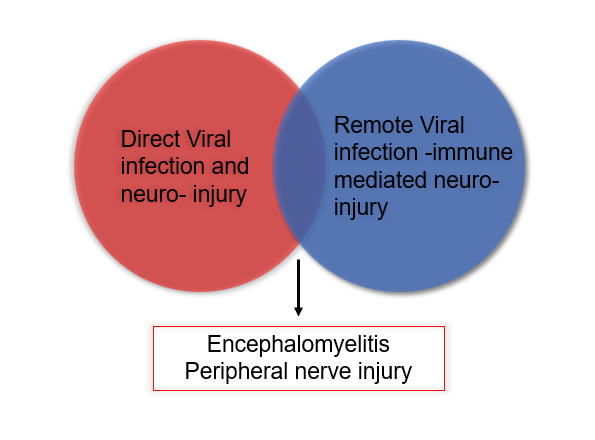
The Germ Theory of Disease was solidified in the 19th century by Louise Pasteur and Robert Koch. They systematically visualized, isolated, and quantified microscopic pathogens as causative agents of diseases and epidemics. Viruses are submicroscopic; therefore, they were discovered later as pathogens by indirect methods [1,2]. Rabies was the first neurological disease determined to be caused by a virus [3,4]. Soon after, the causes of polio and herpes were determined to be viral [5-7]. In the 1930s, viruses were found to be the etiologic agents for the pandemics of arboviral encephalitides such as St. Louis encephalitis and Japanese encephalitis. In the next decade, the viral nature of aseptic meningitis and herpes simplex sporadic encephalitis was discovered [1,4]. From the 1950s through the 1970s, interest in “slow viral diseases” arose as potential models for the nervous system’s chronic diseases, such as multiple sclerosis [8,9]. Acute and chronic viral infections of the nervous system have been extensively documented in the literature [1,10]. However, many of the fulminant and some not so severe neurological injuries are not due to direct infection of neurons by the viruses but due to indirect immune-mediated damage to the nervous system or a bystander effect (Figure 1).
During millions of years of coexistence, a complex system of cohabitation has developed between viruses and their host cells -bacteria to mammals. At different stages of this evolution, flare-ups of destructive infection in the host population occurs that eventually reaches homeostasis between the virus and the host. The 2019 coronavirus disease (COVID-19) pandemic is likely such an inimical phase in the interaction of viral pathogens and the host human population. At first glance, this relationship appears to favor viruses. They need the host cells for survival and replication; viral proteins and the viral envelope are elaborated by the host cells. But if we take a longer view of this symbiosis, perhaps the non-lethal viral infections enrich the host genome and contribute to the molecular evolution. To achieve cohabitation and homeostasis, viruses, including SARS-CoV-2 and the host cells, have developed a complicated chemical dance with the host cell surface molecules.
Viruses require bacterial and eukaryotic cells as hosts, and different types of viruses have evolved to infect and live inside specific types of cells. Therefore, entry into the cells, replication, and exit from the host cells are the necessary steps of the infection cycle. As noted above, there are at least two general mechanisms of injury to the nervous system during viral infection: First, direct invasion of neurons and glia by the virus, and neural damage during viral replication, such as infection with rabies, polio, and herpes viruses [11,12]. In this scenario, the causative agent is isolated from the infected neural tissue, and cell injury/death is associated with viral replication [5]. Second, an immune-mediated process where damage to the nervous system occurs in a temporally and spatially separate setting from the viral infection (Figure 1), partly due to a dysregulated immune system. These include HIV related encephalopathies, autoimmune encephalitis, and Guillain-Barre syndrome. This separation is somewhat artificial as in every infection, both the pathogens and the immune reaction go hand and hand (Figure 1). The current data indicate that neurological injury secondary to SARSCoV- 2 infection falls in the second group.
SARS-CoV-2 Entry into the Host Cells
The family of Coronaviruses is a thriving group of microbial pathogens that infect many species of birds and mammals [13]. SARS-CoV-2 is an RNA virus and belongs to the beta coronavirus genus. It is a zoonotic pathogen native to bats and is likely transmitted to humans via an as yet unknown intermediary [14-16]. Bats and rodents are rich sources of zoonotic viruses because different species of these animals share the same viruses, allowing them to adapt to various hosts without killing them [16-18]. Furthermore, specific groups within a species are responsible for the transmission of the parasite [19]. Once SARS-CoV-2 achieved a hold in the human host, it has made further adaptations to form new clades. The new clades of the virus have accelerated the human-to-human transmission, contributing to the severity of the COVID-19 pandemic [16,20].
Similar to other respiratory pathogens, SARS-CoV-2 is primarily transmitted from human to human by inhalation of virus-containing airborne droplets in crowded, closed quarters. SARS-CoV-2 is a host and cell-specific pathogen, and its cell specificity is determined by the presence of ACE2 on cell membranes [14,21]. Although SARS-CoV-2 carries “respiratory” in its moniker, once inside the organism, it exploits the broadly expressed angiotensinconverting enzyme 2 (ACE2) cell surface molecule for entry into the target cells [22,23]. ACE2 is primarily expressed on the endothelial and epithelial cell membranes [24]. This receptor selectivity contributes to the infection’s specificity and breadth since other organs and cells that express ACE2 are affected [22]. Thus, ACE2 is a cognate cell surface receptor and an essential component of the molecular machinery that holds the key to the entry of SARS-CoV-2 into the host cells. ACE2 also changes the conformation of the spike protein [25] on the viral envelope by a catalytic process that allows the viral envelope to fuse with the host cell membrane [21,26]. The conformational changes are aided by other cell surface enzymes, including transmembrane serine protease2 (TMPRSS2) [27], Furin [28], and ACE2. These proteins are epithelial-specific proteolytic enzymes and cleave a polybasic site at the S1- S2 junction of the spike protein rearranging its structure [25,28,29], allowing the viral envelope to fuse with the target cell membrane.
SARS-CoV-2 and Neurological Damage
Only a small number of patients who test positive for SARS-CoV-2 become ill, and an even smaller number of sick patients experience neurological symptoms and signs [30-35]. The majority of patients with neurological injury had a cerebrovascular or hypoxic event [32,33], and patients who experience neurologic injury during COVID-19 infection have a poor outcome [30,31]. Neurological manifestations of systemic viral infections can include acute and subacute aseptic meningitis, acute and subacute encephalopathy, myelopathy, various forms of Guillain-Barre Syndrome, and myopathy. Since the beginning of the COVID-19 pandemic, a steady stream of reports and reprints cataloging neurological injuries associated with the disease has been published. Most of the articles have been in the form of case reports or case series, adding to the repertoire of clinical information about the COVID-19. Neurological injuries associated with COVID-19 include stroke [36-38], loss of sense of taste and smell [39,40], Guillain-Barre Syndrome [41], upper motor neurons signs with abnormal MRI and EEGs [30-32] as well as neuropsychiatric disorders [32,42]. The mechanisms of neurological manifestations of COVID-19 and its pathogenesis sequence are not fully understood; therefore, understanding the steps that lead to neurological injury in COVID 19 patients provide the opportunity to interfere before the neurological damage occur.
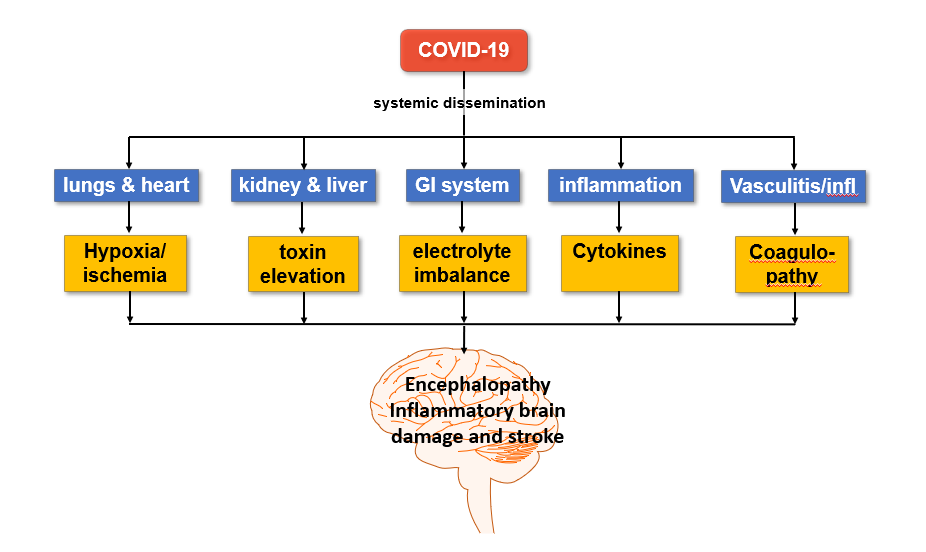
The current data suggest that the neurological injuries secondary to COVID-19 are secondary to systemic infection (Figure 2) rather than a direct invasion of the nervous system by the virus [33,43].
1. A very small number of COVID-19 patients, mostly those who are critically ill, exhibit neurological signs and symptoms.
2. The recent published human neuropathology studies [43] demonstrate low viral levels and no direct brain injury by SARS-CoV-2 in the brain tissue of COVID-19 decedents [43-45]. Only light immunocytochemical staining of viral spike proteins was observed in human brain specimens [44,45], and electron microscope studies show possible clusters of the virus only in the cells close to the surface of the brain [44,45].
3. In experiments that used hACE2-expressing animal models and human brain organoids [45], it was noted that SARS-CoV-2 infects and kills neurons and causes fulminant encephalitis in animals. However, the virulence of SARSCoV- 2 infection is variable within and between species and much depends on the presence of co-morbidities. It is unclear whether the virus can enter in situ neurons in a significant enough number to establish intracellular infection and exit successfully. Questions asked from these experimental models should be about specific steps of viral entry, replication, and exit from the cells and not about the severity of the disease and effects of the virus on the organoids because the organoids are far from the reality of the human infection.
4. ACE2, as the cognate receptor of SARS-CoV-2, is essential for viral entry into cells. Neurons and glia do not express ACE2; however, vascular endothelial cells are rich in ACE2 [24]. In areas that the blood-brain barrier is naturally breached, the virus can penetrate the brain, but no fulminant encephalitis has been observed [43].
5. Host immune failure contributes significantly to the virulence of COVID-19 and its neurological manifestations. Immune incompetence can be due to failure of antigen recognition, generalized immune impairment due to viral infection [46], specific genetic factors [47], and immune failure secondary to other conditions such as obesity [48]. In the nervous system, innate immunity (microglia and astrocytes) plays an essential role in fighting infections because of the blood-brain barrier. No data is demonstrating that the innate brain immunity is affected by the COVID-19.
6. Finally, immune dysregulation can contribute to neurological damage and injury. Immune dysregulation is different from immune failure in that parts of immune system are active. Encephalitis and encephalopathy can be caused by the interaction between the host immune system and the virus, causing an unfavorable neurological outcome, such as in limbic encephalitis and other autoimmune types encephalitides [49], Guillain-Barre Syndrome, and the so-called “slow virus” diseases.
At this juncture, enough molecular and organizational knowledge is available from different disciplines to infer some of the mechanisms of injury to human nervous systems by SARS-CoV-2 that can help diagnose and treat this disease’s neurological manifestation. SARS-CoV-2 enters the host cells using the cognate receptor ACE2. ACE2 is primarily expressed on epithelial/endothelial cells. Thus, our approach to neurological manifestations of COVID-19 must consider this fact. Loss of sense of taste and smell is an initial symptom of COVID-19 because the virus enters the nasal and oral cavities first, and the epithelial cells are the receptors for these senses. COVID-19 associated stroke occurs because of likely injury to vascular endothelial cells and coagulopathy that then cause thrombo-embolism. Encephalopathy in acute and convalescent COVDID-19 patients [50] is multifactorial and likely secondary to hypoxia as well as metabolic and immunological abnormalities (Figure 2). It appears that neurologic injuries in COVID-19 patients are likely indirect and a ‘bystander’ injury since SARS-CoV-2 does not have tropism for the neural cells and tissues.
References
2. Amoss HL, Eberson F. Therapeutic experiments with Rosenow’s antipoliomyelitic serum. The Journal of Experimental Medicine. 1918 Feb 1;27(2):309-17.
3. Watson EM. The negri bodies in rabies. The Journal of Experimental Medicine. 1913 Jan 1;17(1):29.
4. Goodpasture EW. A study of rabies, with reference to a neural transmission of the virus in rabbits, and the structure and significance of Negri bodies. The American Journal of Pathology. 1925 Nov;1(6):547.
5. Parker Jr F, Nye RN. Studies on filterable viruses: II. Cultivation of herpes virus. The American Journal of Pathology. 1925 May;1(3):337.
6. Cockayne EA. Poliomyelitis and Polio-Encephalitis. Postgraduate Medical Journal. 1927 Jul;2(22):151.
7. Richards M. Case of Polio-encephalomyelitis. Proceedings of the Royal Society of Medicine. 1915;8(Sect Study Dis Child):85-6.
8. Tarlinton RE, Martynova E, Rizvanov AA, Khaiboullina S, Verma S. Role of Viruses in the Pathogenesis of Multiple Sclerosis. Viruses. 2020 Jun;12(6):643.
9. Tselis A. Evidence for viral etiology of multiple sclerosis. InSeminars in Neurology. 2011 Jul;31(3):307-16.
10. Booss J, Dann PR, Griffith BP, Kim JH. Host defense response to cytomegalovirus in the central nervous system. Predominance of the monocyte. The American Journal of pathology. 1989 Jan;134(1):71.
11. Jackson AC. Rabies pathogenesis. Journal of Neurovirology. 2002 Aug;8(4):267-9.
12. Lafon M. Modulation of the immune response in the nervous system by rabies virus. Current Topics in Microbiology and Immunology. 2005;289:239-58.
13. Poutanen SM (2018) Human Coronaviruses. In Long S (ed) Principle and practice of pediatric infectious Diseases pp. 1148-1152.
14. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. The Lancet. 2020 Feb 22;395(10224):565-74.
15. Luis AD, Hayman DT, O’Shea TJ, Cryan PM, Gilbert AT, Pulliam JR, et al. A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special?. Proceedings of the Royal Society B: Biological Sciences. 2013 Apr 7;280(1756):20122753.
16. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nature Medicine. 2020 Apr;26(4):450-2.
17. Luis AD, O’Shea TJ, Hayman DT, Wood JL, Cunningham AA, Gilbert AT, et al. Network analysis of host–virus communities in bats and rodents reveals determinants of cross-species transmission. Ecology Letters. 2015 Nov;18(11):1153-62.
18. Streicker DG, Gilbert AT. Contextualizing bats as viral reservoirs. Science. 2020 Oct 9;370(6513):172-173.
19. Streicker DG, Fenton A, Pedersen AB. Differential sources of host species heterogeneity influence the transmission and control of multihost parasites. Ecology Letters. 2013 Aug;16(8):975-84.
20. Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. Tracking changes in SARSCoV- 2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020 Aug 20;182(4):812-27.
21. Heald-Sargent T, Gallagher T. Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses. 2012 Apr;4(4):557-80.
22. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003 Nov;426(6965):450-4.
23. Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, et al. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020 May;581(7807):221-4.
24. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis GV, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland. 2004 Jun;203(2):631-7.
25. Turonová B, Sikora M, Schürmann C, Hagen WJ, Welsch S, Blanc FE, et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science. 2020 Oct 9;370(6513):203-8.
26. Belouzard S, Millet JK, Licitra BN, Whittaker GR. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses. 2012 Jun;4(6):1011-33.
27. Stopsack KH, Mucci LA, Antonarakis ES, Nelson PS, Kantoff PW. TMPRSS2 and COVID-19: Serendipity or Opportunity for Intervention?. Cancer Discovery. 2020 Jun 1;10(6):779-82.
28. Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nature reviews Molecular Cell Biology. 2002 Oct;3(10):753-66.
29. Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, et al. Cell entry mechanisms of SARS-CoV-2. Proceedings of the National Academy of Sciences. 2020 May 26;117(21):11727-34.
30. Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurology. 2020 Jun 1;77(6):683-90.
31. Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, et al. Neurologic features in severe SARS-CoV-2 infection. New England Journal of Medicine. 2020 Apr 15; 382:2268-2270.
32. Varatharaj A, Thomas N, Ellul MA, Davies NW, Pollak TA, Tenorio EL, et al. Neurological and neuropsychiatric complications of COVID-19 in 153 patients: a UK-wide surveillance study. The Lancet Psychiatry. 2020 Oct 1;7(10):875-82.
33. Paterson RW, Brown RL, Benjamin L, Nortley R, Wiethoff S, Bharucha T, Jayaseelan DL, Kumar G, Raftopoulos RE, Zambreanu L, Vivekanandam V. The emerging spectrum of COVID-19 neurology: clinical, radiological and laboratory findings. Brain. 2020 Oct;143(10):3104-20.
34. Ellul MA, Benjamin L, Singh B, Lant S, Michael BD, et al. Neurological associations of COVID-19. Lancet Neurol. 2020 Sep;19(9):767-783.
35. Koralnik IJ, Tyler KL. COVID-19: A Global Threat to the Nervous System. Annals of Neurology. 2020 Jul;88(1):1-11.
36. Yaghi S, Ishida K, Torres J, Mac Grory B, Raz E, Humbert K,et al. (2020) SARS-CoV-2 and Stroke in a New York Healthcare System. Stroke. 2020 Jul;51(7):2002- 2011.
37. Oxley TJ, Mocco J, Majidi S, Kellner CP, Shoirah H, Singh IP, De Leacy RA, Shigematsu T, Ladner TR, Yaeger KA, Skliut M. Large-vessel stroke as a presenting feature of Covid-19 in the young. New England Journal of Medicine. 2020 May 14;382(20):e60.
38. Spence JD, De Freitas GR, Pettigrew LC, Ay H, Liebeskind DS, Kase CS, et al. Mechanisms of stroke in COVID-19. Cerebrovascular Diseases. 2020;49(4):451-8.
39. Menni C, Valdes AM, Freidin MB, Sudre CH, Nguyen LH, Drew DA, et al. Real-time tracking of selfreported symptoms to predict potential COVID-19. Nature Medicine. 2020 May 11:1-4.
40. Cooper KW, Brann DH, Farruggia MC, Bhutani S, Pellegrino R, Tsukahara T, et al. COVID-19 and the chemical senses: supporting players take center stage. Neuron. 2020 Jul 22; 107(2): 219–233.
41. Toscano G, Palmerini F, Ravaglia S, Ruiz L, Invernizzi P, Cuzzoni MG, et al. Guillain–Barré syndrome associated with SARS-CoV-2. New England Journal of Medicine. 2020 Jun 25;382(26):2574-2576.
42. Rogers JP, Chesney E, Oliver D, Pollak TA, McGuire P, Fusar-Poli P, et al. Psychiatric and neuropsychiatric presentations associated with severe coronavirus infections: a systematic review and meta-analysis with comparison to the COVID-19 pandemic. Lancet Psychiatry. 2020 Jul;7(7):611-627.
43. Matschke J, Lütgehetmann M, Hagel C, Sperhake JP, Schröder AS, Edler C, et al. Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. The Lancet Neurology. 2020 Nov 1;19(11):919-29.
44. Solomon IH, Normandin E, Bhattacharyya S, Mukerji SS, Keller K, Ali AS, et al. Neuropathological Features of Covid-19. New England Journal of Medicine. 2020 Sep 3;383(10):989-992.
45. Song E, Zhang C, Israelow B, Lu-Culligan A, Prado AV, et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. bioRxiv [Preprint]. 2020 Sep 8:2020.06.25.169946.
46. Schub D, Klemis V, Schneitler S, Mihm J, Lepper PM, Wilkens H, et al. High levels of SARS-CoV-2–specific T cells with restricted functionality in severe courses of COVID-19. JCI Insight. 2020 Oct 15;5(20).
47. Ellinghaus D, Degenhardt F, Bujanda L, Buti M, Albillos A, Invernizzi P, et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. The New England Journal of Medicine. 2020 Jun 17:NEJMoa2020283.
48. Andersen CJ, Murphy KE, Fernandez ML. Impact of Obesity and Metabolic Syndrome on Immunity. Advances in Nutrition. 2016;7(1):66-75.
49. Ellul MA, Wood G, Tooren HVD, Easton A, Babu A, Michael BD. Update on the diagnosis and management of autoimmune encephalitis. Clinical Medicine (Lond). 2020 Jul;20(4):389-392.
50. Kotfis K, Williams Roberson S, Wilson JE, Dabrowski W, Pun BT, Ely EW. COVID-19: ICU delirium management during SARS-CoV-2 pandemic. Critical care. 2020 Dec;24:1-9.