Abstract
Neurocysticercosis (NC) presents a spectrum of clinical symptoms, with two broad clinical entities based on the CNS location of the parasite: parenchymal (P-NC) or extraparenchymal (EP-NC). In view of the importance of inflammation in the pathogenesis of NC, it is surprising that the possible occurrence of anti-brain autoantibodies in NC has not been explored until recently. In this study the presence of autoantibodies to nine ubiquitous intracellular proteins from extraparenchymal (EP-NC) was reported. Levels of these autoantibodies were greatly reduced or absent in P-NC, an observation consistent with our understanding of the immunological potential of these two distinct anatomical areas of the brain. In more recent work, we similarly observed autoantibodies to tubulin and MOG in the CSF of EP-NC, but not P-NC patients. In addition to the evident importance of the early inflammatory response, the identification of autoantibodies to the neuronal surface protein MOG (and perhaps other anti-neuronal autoantibodies) provides a potentially critical role in the pathogenesis of NC through their predicted potential for antibody mediated cytotoxicity and the subsequent release of intracellular proteins which, in turn, would stimulate a cascade of more autoantibodies to the liberated intracellular proteins. These, together with antibodies to metacestode proteins may be expected to play an important role in the continuously evolving and variable pathogenesis of NC, as is summarized in the hypothetical model presented.
Keywords
Neurocysticercosis; Anti-brain autoantibodies; Tubulin; MOG; Extraparenchymal; Parenchymal
Commentary
Neurological diseases are a major cause of disability and the second cause of death today [1]. This reality has stimulated the search for predictive biomarkers facilitating early diagnosis and the design of appropriate treatments. One source of such biomarkers for inflammatory neuropathologies is autoantibodies. Indeed, of the 5.7% of the world’s population with autoantibodies, 3% have been demonstrated to be anti-brain autoantibodies associatedy with a wide range of nervous system diseases, typically in individuals with Blood-Brain Barrier disruption [2].
In addition to non-infectious diseases, previous studies have suggested that parasites may also trigger activation of both cellular and serological anti-brain autoimmune mechanisms [3]. Such autoantibodies have been explained by (1) Molecular mimicry between host and parasite molecules [3-5] and (2) The release of parasite immunogenic antigens normally “invisible” to the immune system directly or through the induction of an exacerbated inflammatory response mediated cell lysis. The relative contribution of these two mechanisms to various autoimmune neurological diseases requires further studies [6]. A crucial question for the future is to define mechanisms of molecular mimicry that exist between Taenia solium and neuronal antigens. At present, the only relevant observations are tenuous: (1) “Glycan mimicry” in many helminths, including Echinococcus granulosus and T. solium [7], (2) 94 tapeworm protein sequences with bioinformatic similarities to stickleback proteins [8] and (3) False positive ELISA’s for E. granulosus with patients presenting with an undifferentiated embryonic sarcoma of the liver, interpreted as evidence for the molecular mimicry hypothesis [9].
Neurocysticercosis (NC), infection of the human Central Nervous System (CNS) by the larval phase of the cestode parasite T. solium, is a neglected zoonotic [10] and usually poverty-related disease of high public health importance that is still a cause of unacceptable morbidity and mortality, not only in endemic lower-income Latin America countries, Africa, and Asia, but also increasingly in high-income countries due to migration [11,12]. As the most common parasite of the CNS worldwide [13], and a major cause of seizures in endemic countries [14], NC is not a single entity. It is a “spectral” disease covering a wide range of clinical symptoms, ranging from benign to frankly life threatening, depending on the number, localization and physiological state of the parasite and the corresponding host inflammatory response. Operationally, NC presents two broad clinical entities reflecting the CNS location of the parasite; parenchymal (P-NC) and extraparenchymal (EP-NC). When the parasite is located in the cerebral parenchyma or in the sulci, the clinical picture is relatively benign and in most cases the parasites are destroyed without major symptoms. The extraparenchymal localization of the parasite in the subarachnoid space or the ventricles is clearly associated to the most severe clinical manifestation; intracranial hypertension caused by hydrocephalus as a result of mechanical obstruction of cerebrospinal fluid (CSF) flow. These patients were more likely to require a ventriculoperitoneal shunt to prevent severe complications from intracranial hypertension resulting from exacerbated inflammation [15]. EP-NC frequently required high doses of intravenous glucocorticoids, to reach therapeutic doses in the CNS and ultimately control neuroinflammation. Indeed, the cysticidal treatment must be administered in conjunction with high doses of glucocorticoids that have to be sustained both during and after the duration of the cysticidal treatment. In view of the importance of inflammation in the pathogenesis of NC, it is therefore surprising that the possible occurrence of anti-brain autoantibodies in the NC has not been explored until this year [16].
In this first study the presence of autoantibodies to brain proteins in CSF from extraparenchymal (EPNC), but not parenchymal (P-NC) patients was reported using quantitative immunoblot methodology. There was striking correlation between the level of autoantibodies and the levels of the secreted metacestode glycoprotein HP-10, suggesting that the level of stimulation of the autoantibody response may be a function of the number of viable parasites. Examination of the immunoblot profiles of the EP-NC CSF samples revealed considerable heterogeneity between them. However, a total of nine proteins were identified by mass spectroscopy in at least 60% of the CSF samples from cases of EP-NC and thus may be provisionally considered to be possible common auto-antigens and worthy of further investigation. Gene-ontology enrichment revealed these 9 proteins to be ubiquitous intracellular proteins (Clusterin (CLU), Transthyretin (TTR), Haptoglobin (HP), Ceruloplasmin (CP), Alpha-2-Macroglobulin (A2M), CARD9 (CARD9), Cytostatin C (CST3), Angiotensin (AGT), 60S ribosomal protein L27a (RP27A)), with the majority secreted and/ or involved in the immune effector process or the defense response.
Similarly, a recent study in multiple sclerosis patients found the targets of these antibodies to be ubiquitous intracellular proteins rather than brain-specific selfantigens, suggesting a nonspecific secondary response, for example to damaged/dead cells, rather than a direct pathogenic involvement [17]. Other examples of autoantibodies to ubiquitous intracellular proteins associated with cerebral disease include autism [18] and pesticide induced neurotoxicity [19].
In contrast, other authors suggest direct pathogenic effect of the CSF anti-brain antibodies in other infectious and non-infectious diseases of the brain. For example, autoantibodies to CNS neuronal surface antigens have been described in association with autoimmune encephalopathies which notably feature psychiatric in addition to neurological symptoms [20,21]. Similarly, in systemic lupus erythematosus, antibodies to double stranded DNA that cross react with the neuronal N-methyl– aspartate receptor, have recently been linked to neurocognitive dysfunction [22,23]. Infection of the CNS with herpes simplex virus can trigger anti-CNS autoimmunity associated with anti-GABA antibodies that can modify the normal activity of this neurotransmissor [24]. Anti-neural autoantibodies have been implicated in the pathogenesis of nerve damage in leprosy patients. Thus, leprosy patients are more likely to be seropositive for peripheral auto-antibodies against spinal cord than normal controls (25% vs 7%). Later on, autoantibodies against intracellular cerebroside sulphate (sulphatide) were also reported and their participation has been proposed to be involved in pathology during periods of inflammation, particularly in multibacillary patients [25].
Clearly, autoantibodies to ubiquitous intracellular proteins would only occur after autoanti-brain antibody mediated or brain injury induced cell death, often in the context of blood-brain barrier disruption and the impact of the host inflammatory response. The consequent damage in the CNS would release intracellular host proteins that could stimulate autoantibody synthesis in peripheral lymph nodes. The resulting autoantibodies in the peripheral blood could then return to the brain through the circumventricular organs or the choroid plexus, structures in the brain linked to the ventricular system and lacking a blood-brain barrier [26]. An additional possibility, facilitated by the initial inflammatory response to the extraparenchymal metacestodes, is the intrathecal synthesis of autoantibodies.
Finally, antibodies to metacestode proteins, released because of inflammation mediated damage would also be similarly generated. These, together with autoantibodies to neuronal cell surface and intracellular proteins, would be predicted to influence the progressive pathogenesis of NC
Our failure to demonstrate autoantibodies to neural cell surface proteins in this first study stimulated a more direct approach, the ELISA examination of Mexican CSF samples with clinically defined cases of EP-NC and P-NC for autoantibodies to the Major Oligodendrocyte glycoprotein surface protein (MOG) and the ubiquitous intracellular protein tubulin (Parkhouse et al., unpublished work). In this study, we also observed the presence of similar and significant levels of autoantibodies recognizing both tubulin and MOG in the CSF from some, but not all, cases of EP-NC. In contrast, such autoantibodies were greatly reduced or absent in the CSF from patients with P-NC, once again an observation consistent with differences in the immunobiology of the extraparenchymal versus parenchymal compartments. Specifically, extraparenchymal cysts are surrounded by CSF, which interacts with the peripheral immune system through its afferent and efferent connections through the dural sinuses to the deep cervical lymph nodes [27]. This communication with the peripheral immune system allowing immune-cell trafficking is much more efficient for this compartment than for the parenchymal one [28]. Moreover, most patients with parenchymal NC do not have an inflammatory CSF. Thus, it is highly relevant that we observed a statistically significant correlation between the number of cells in the CSF and the autoantibody response to MOG (R=0.68; P=0.0001) and to tubulin (R=0.72; P=0.0001) in patients with extraparenchymal NC. CSF protein titers were also significantly associated with autoantibodies titers. (R= 0.51, P=0.008 for tubulin, and R=0.48, P=0.012 for MOG). Moreover, CSF proteins closely correlate with CSF cellularity. Thus, we decided to report the relation of autoantibodies with CSF cellularity as it is a parameter more clearly involved with acute inflammation and more frequently used for this purpose.
Unfortunately, we included only 5 patients with parenchymal NCC, 4 of them with multiple parasites. In extraparenchymal, all the patients included presented parasites in the subarachnoid space, and all but one has multiple parasites. In this situation, it was impossible to make any statistical analysis. It is clear that complementary studies with more patients are necessary to better understand the relation between autoantibodies and pathogenicity.
Both tubulin and MOG have been recognized as targets for autoantibodies in a variety of cerebral neuropathologies. Examples of autoantibodies to ubiquitous intracellular proteins such as tubulin include Behcet’s Disease [29], Systemic Lupus Erythematosus [30] and Sydenham’s Chorea [31]. Significantly, intrathecally synthesized antitubulin autoantibodies have also been identified as a potential risk factor in the latter three reports and appear to be related to axonal damage [32]. While antibodies to MOG were originally thought to be involved in multiple sclerosis, over the last decade, a robust association of autoantibodies to MOG with optic neuritis, brainstem encephalitis, and acute disseminated encephalomyelitis has been demonstrated. However recent reports consider MOG-associated a disease entity in its own right, immunopathogenetically distinct from classic multiple sclerosis and neuromyelitis optica spectrum disorders [33]. The extent of CNS inflammatory diseases has recently broadened to include a new condition associated with pathogenic serum antibodies against MOG, which has been recognized as a distinct clinical disorder with a different treatment [34,35]. Indeed, new studies encountered patients with seizures associated with MOGIgG disease [36].
From the practical point of view, autoantibodies in NC may provide novel strategies for the management and therapy of NC; for example, autoantibodies in NC patients might serve as a “biomarker” indicating the intensity of inflammation, which is a crucial factor to consider for the design of the anti-inflammatory therapeutic scheme associated to cysticidal drugs for EP-NC. EP-NC frequently required high doses of intravenous glucocorticoids, to reach therapeutic doses in the CNS and ultimately control neuroinflammation. Indeed, in EP-NC high doses of glucocorticoids must be administered both during and after the duration of the cysticidal treatment.
Furthermore, this work raises two other rational clinical possibilities: immunosuppressive therapy and the blocking of potentially pathogenic autoantibodies and other proinflammatory factors through the administration of normal immunoglobulin, as indeed has been reported in cases of lupus erythematosus [37] and various neurological, in particular neuro-inflammatory, conditions, such as, intractable autoimmune epilepsy, paraneoplastic syndrome, and autoimmune encephalitis [38,39], as well as the administration of monoclonal antibodies for the therapy of emerging infectious diseases [40].
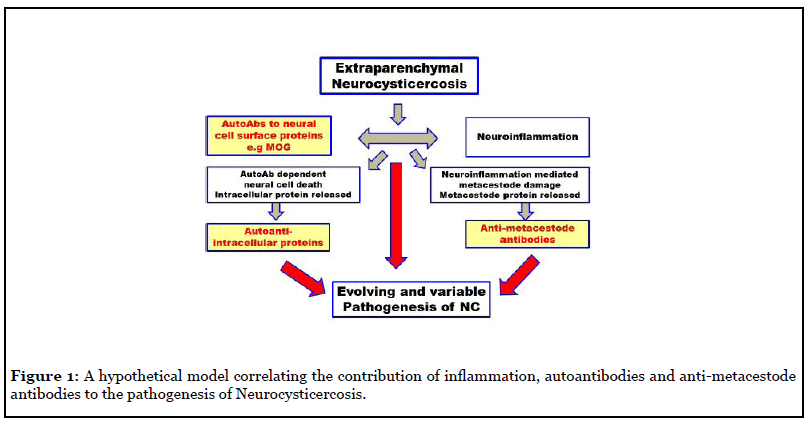
In conclusion, the finding of significant levels of autoantibodies in the CSF of EP-NC, but not P-NC, patients is consistent with our understanding of the immunological potential of these two distinct anatomical areas of the brain, and highlights the need for further studies explaining the relationships between autoimmunity, neuro-inflammation and NC. In addition to the evident importance of the early inflammatory response, the identification of autoantibodies to the neuronal surface protein MOG (and perhaps other anti-neuronal autoantibodies) provides a potentially critical role through their predicted antibody mediated cytotoxicity mediated liberation of intracellular proteins which, in turn, would stimulate a cascade of more autoantibodies to the liberated intracellular proteins. These autoantibodies, together with antibodies to metacestode proteins, may be expected to play an important role in the continuously evolving and variable pathogenesis of neurocysticercosis, as is summarized in the hypothetical model presented in Figure 1.
References
2. Diamond B, Honig G, Mader S, Brimberg L, Volpe BT. Brain-reactive antibodies and disease. Annual Review of Immunology. 2013 Mar 21;31:345-85.
3. Rojas M, Restrepo-Jiménez P, Monsalve DM, Pacheco Y, Acosta-Ampudia Y, Ramírez-Santana C, et al. Molecular mimicry and autoimmunity. Journal of Autoimmunity. 2018 Dec 1;95:100-23.
4. Abu-Shakra M, Buskila D, Shoenfeld Y. Molecular mimicry between host and pathogen: examples from parasites and implication. Immunology Letters. 1999 Apr 1;67(2):147-52.
5. Nestor J, Gata-Garcia A, Arinuma Y, Fujieda Y, Kowal C, Diamond B. Immune-mediated brain pathology: from autoantibodies to microglia. Discovery Medicine. 2016 Oct 24;22(121):201-7.
6. Klein RS, Garber C, Howard N. Infectious immunity in the central nervous system and brain function. Nature immunology. 2017 Feb;18(2):132-41.
7. Van Die I, Cummings RD. Glycan gimmickry by parasitic helminths: a strategy for modulating the host immune response?. Glycobiology. 2010 Jan 1;20(1):2-12.
8. Hebert FO, Phelps L, Samonte I, Panchal M, Grambauer S, Barber I, et al. Identification of candidate mimicry proteins involved in parasite-driven phenotypic changes. Parasites & Vectors. 2015 Dec 1;8(1):225.
9. Letherer A, Mastenbrook J, VanEnk RA, Bauler LD. Undifferentiated Embryonal Sarcoma of the Liver Presents as a Molecular Mimic of Parasitic Infection. Cureus. 2020 Jan;12(1).
10. FAO W. Multicriteria-based ranking for risk management of food-borne parasites. Rome: Food and Agriculture Organization. 2014.
11. Symeonidou I, Arsenopoulos K, Tzilves D, Soba B, Gabriël S, Papadopoulos E. Human taeniasis/cysticercosis: a potentially emerging parasitic disease in Europe. Annals of Gastroenterology. 2018 Jul;31(4):406.
12. O’Neal SE, Flecker RH. Hospitalization frequency and charges for neurocysticercosis, United States, 2003–2012. Emerging Infectious Diseases. 2015 Jun;21(6):969.
13. Carpio A, Fleury A, Romo ML, Abraham R. Neurocysticercosis: the good, the bad, and the missing. Expert Review of Neurotherapeutics. 2018 Apr 3;18(4):289-301.
14. Tellez-Zenteno JF, Hernandez-Ronquillo L. Epidemiology of neurocysticercosis and epilepsy, is everything described?. Epilepsy & Behavior. 2017 Nov 1;76:146-50.
15. Hamamoto Filho PT, Zanini MA, Fleury A. Hydrocephalus in neurocysticercosis: challenges for clinical practice and basic research perspectives. World Neurosurgery. 2019 Jun 1;126:264-71.
16. Parkhouse RM, Carpio A, Cortez MM, von Kriegsheim A, Fesel C. Anti-brain protein autoantibodies are detectable in extraparenchymal but not parenchymal neurocysticercosis. Journal of Neuroimmunology. 2020 Apr 6:577234.
17. Brändle SM, Obermeier B, Senel M, Bruder J, Mentele R, Khademi M, et al. Distinct oligoclonal band antibodies in multiple sclerosis recognize ubiquitous self-proteins. Proceedings of the National Academy of Sciences. 2016 Jul 12;113(28):7864-9.
18. Frye RE, Sequeira JM, Quadros EV, James SJ, Rossignol DA. Cerebral folate receptor autoantibodies in autism spectrum disorder. Molecular Psychiatry. 2013 Mar;18(3):369-81.
19. Abd El Rahman HA, Salama M, El-Hak SA, El- Harouny MA, ElKafrawy P, Abou-Donia MB. A panel of autoantibodies against neural proteins as peripheral biomarker for pesticide-induced neurotoxicity. Neurotoxicity Research. 2018 Feb 1;33(2):316-36.
20. Armangue T, Leypoldt F, Dalmau J. Auto-immune encephalitis as differential diagnosis of infectious encephalitis. Current Opinion in Neurology. 2014 Jun;27(3):361.
21. Pollak TA, Beck K, Irani SR, Howes OD, David AS, McGuire PK. Autoantibodies to central nervous system neuronal surface antigens: psychiatric symptoms and psychopharmacological implications. Psychopharmacology. 2016 May 1;233(9):1605-21.
22. Mader S, Brimberg L, Diamond B. The role of brainreactive autoantibodies in brain pathology and cognitive impairment. Frontiers in Immunology. 2017 Sep 11;8:1101.
23. Arinuma Y. Antibodies and the brain: anti-N-methyl- D-aspartate receptor antibody and the clinical effects in patients with systemic lupus erythematosus. Current Opinion in Neurology. 2018 Jun 1;31(3):294-9.
24. Alexopoulos H, Akrivou S, Mastroyanni S, Antonopoulou M, Dinopoulos A, Giorgi M, et al. Postherpes simplex encephalitis: a case series of viral-triggered autoimmunity, synaptic autoantibodies and response to therapy. Therapeutic Advances in Neurological Disorders. 2018 Apr 19;11:1756286418768778.
25. Wheeler PR, Raynes JG, McAdam KP. Autoantibodies to cerebroside sulphate (sulphatide) in leprosy. Clinical & Experimental Immunology. 1994 Oct;98(1):145-50.
26. Marques F, Sousa JC, Brito MA, Pahnke J, Santos C, Correia-Neves M, Palha JA. The choroid plexus in health and in disease: dialogues into and out of the brain. Neurobiology of Disease. 2017 Nov 1;107:32-40.
27. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015 Jul;523(7560):337-41.
28. Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nature Immunology. 2017 Feb;18(2):123.
29. Cheng Y, Zhao X, Chen Y, Li Y, Jia R, Zhu L, et al. Circulating immune complexome analysis identified antitubulin- a-1c as an inflammation associated autoantibody with promising diagnostic value for Behcet’s Disease. PloS one. 2018 Jun 14;13(6):e0199047.
30. Shaban A, Leira EC. Neurological Complications in Patients with Systemic Lupus Erythematosus. Current neurology and neuroscience reports. 2019 Dec 1;19(12):97.
31. Waheed W, Boyd J, Khan F, Mount SL, Borden NM, Tandan R. Double trouble: para-neoplastic anti-PCA-2 and CRMP-5-mediated small fibre neuropathy followed by chorea associated with small cell lung cancer and evolving radiological features. Case Reports. 2016 Aug 29;2016:bcr2016215158.
32. Švarcová J, Fialova L, Bartoš A, Šteinbachová M, Malbohan I. Cerebrospinal fluid antibodies to tubulin are elevated in the patients with multiple sclerosis. European Journal of Neurology. 2008 Nov;15(11):1173-9.
33. Jarius S, Paul F, Aktas O, Asgari N, Dale RC, De Seze J, et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. Journal of Neuroinflammation. 2018 Dec 1;15(1):134.
34. Jurynczyk M, Jacob A, Fujihara K, Palace J. Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease: practical considerations. Practical Neurology. 2019 Jun 1;19(3):187-95.
35. Wynford-Thomas R, Jacob A, Tomassini V. Neurological update: MOG antibody disease. Journal of Neurology. 2019 May 1;266(5):1280-6.
36. Hamid SH, Whittam D, Saviour M, Alorainy A, Mutch K, Linaker S, et al. Seizures and encephalitis in myelin oligodendrocyte glycoprotein IgG disease vs aquaporin 4 IgG disease. JAMA Neurology. 2018 Jan 1;75(1):65-71.
37. Yildirim-Toruner C, Diamond B. Current and novel therapeutics in the treatment of systemic lupus erythematosus. Journal of Allergy and Clinical Immunology. 2011 Feb 1;127(2):303-12.
38. Uchibori A, Chiba A. 2015 Autoantibodies in Guilliain- Barre Syndrome. Brain Nerve. 67:1347-57 DOI 10.11477/ mf1416200305.
39. Chen Y, Wang C, Xu F, Ming F, Zhang H. Efficacy and tolerability of intravenous immunoglobulin and subcutaneous immunoglobulin in neurologic diseases. Clinical Therapeutics. 2019 Oct 1;41(10):2112-36.
40. Marston HD, Paules CI, Fauci AS. Monoclonal antibodies for emerging infectious diseases—borrowing from history. N Engl J Med. 2018 Apr 19;378(16):1469-72.