Abstract
Introduction: Alzheimer’s disease (AD) is the most common cause of dementia globally and imposes a growing burden on healthcare systems.
Historical background: Alois Alzheimer reported the first case of AD in 1907, describing “particular changes in cortical cell clusters” on brain biopsy and attributing the patient’s behavioral changes to these lesions.
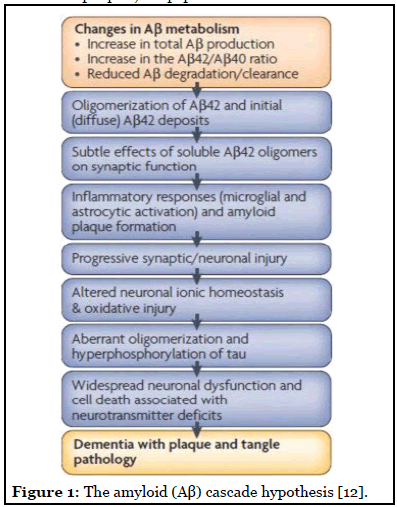
Pathogenesis: AD is a complex, multifactorial, neurodegenerative disease implicating the interactions of one’s genetic makeup, education, age, and environment. Currently the most accepted theory for the development of AD is the amyloid cascade hypothesis, which attributes clinical signs/symptoms to the overwhelming presence of amyloid beta (Ab) peptides, leading to increased deposition into amyloid plaques and the eventual result of neuronal damage.
Presentation: The most common presentation of those with AD involves an elderly individual with gradual decline in memory centered cognitive decline.
Testing: Though AD remains a clinical diagnosis, imaging such as fluorodeoxyglucose positron emission tomography (PET) and amyloid PET and biomarkers in cerebrospinal fluid (CSF) can be helpful in evaluating some patients.
Histology: The histology of AD is composed primarily of extracellular amyloid plaques consisting of misfolded Ab peptides and intracellular neurofibrillary tangles consisting of hyperphosphorylated tau. Eventually, these lead to gross anatomical findings of atrophy diffusely.
Treatment: Current pharmaceutical treatment available for AD include cholinesterase inhibitors as well as memantine. Other aims include increasing one’s cognitive reserve and providing a nutritional approach to prevent or slow the progression of disease.
Future Directions: Most therapeutics in development are intended to achieve disease modification by targeting amyloid plaques or neurofibrillary tangles of tau. There is increasing focus on identifying and prophylactically treating patients with preclinical AD and individuals with risk factors for cognitive decline.
Introduction
The worldwide prevalence of dementia is estimated to be over 45 million people. Alzheimer’s disease (AD) is the most common cause of dementia, responsible for 60-80% of cases [1]. An estimated 5.8 million Americans ages 65 and older have AD, a number which could grow to 13.8 million by 2050. As the number of people suffering from AD increases, so does the economic burden of care. Payments for healthcare and hospice services for Americans 65 and older with dementia are estimated to be $305 billion in 2020 [2].
The neuropathology of AD consists of extracellular betaamyloid plaque depositions and intracellular neurofibrillary tangles of hyperphosphorylated tau. AD remains a clinical diagnosis, although cerebrospinal fluid (CSF) and positron emission tomography (PET) biomarkers can increase diagnostic accuracy [3]. Current treatments, including cholinesterase inhibitors and memantine, improve quality of life but do not modify or slow the disease course. Current research aims to treat underlying pathology of active AD as well as identify and stage interventions in those with preclinical, or asymptomatic, AD.
Historical Background
Alois Alzheimer, a German physician, reported the first case of Alzheimer’s disease in 1907 [4]. He first saw Auguste Deter, a 51-year-old woman, in 1901. Auguste’s husband Karl brought her to a mental hospital after she began exhibiting unusual behavior, including hiding items, threatening neighbors, and accusing her husband of adultery. She also lost the ability to do daily activities such as cooking and housework. Auguste came under Alzheimer’s care at a mental hospital in Frankfurt. There he observed and recorded her behavioral patterns: she could speak but not write her own name, she could name objects such as a pencil but not the food she was eating, she was polite sometimes but loud and offensive at other times. He diagnosed Auguste with “presenile dementia” [5].
Upon her death in 1906, Alzheimer’s biopsy of her brain revealed diffuse cortical atrophy and “particular changes in cortical cell clusters” [6]. Alzheimer described plaques and tangles of nerve fibers which researchers would identify in the 1980’s as beta amyloid plaques and neurofibrillary tangles of tau [7,8]. That year, Alzheimer gave a presentation on Auguste at a German psychiatry conference, asserting these cortical lesions to be the cause of her symptoms. He published a research paper the next year, and a psychiatry textbook in 1910 named the disorder ‘Alzheimer’s disease.’
The clinical diagnostic criteria for AD were standardized in the U.S. in 1984 [9]. They were revised in 2011 and 2018 to create separate diagnoses for the preclinical, mild cognitive impairment (MCI) and dementia stages of AD and to recognize the role of biomarkers in AD diagnosis [10,11].
Pathogenesis
AD is a complex, multifactorial, neurodegenerative disease, resulting from complicated interactions of one’s genetic makeup, education, age, and environment. Many hypotheses have laid the foundation to gain understanding of the etiology of the disease, with one of the oldest being the cholinergic hypothesis. This hypothesis is based upon the fact that AD patients show reduction in activity of choline acetyltransferase and acetylcholinesterase in the cerebral cortex compared with the normal brain [12]. Postmortem brain tissue from patients with AD confirmed the reduced neurotransmitter pathway activity, revealing that degeneration of cholinergic neurons and loss of cholinergic neurotransmission significantly contributes to the cognitive impairment seen in those with AD [12]. The Tau hypothesis has also been proposed, considering AD histopathology reveals intraneuronal neurofibrillary lesions made up of tau proteins. Tau proteins are mainly found in neurons and are involved in the assembly and stabilization of the neuronal microtubule network. Tau protein becomes pathological when the phosphorylation regulation becomes unchecked and hyperphosphorylated tau proteins polymerize into filaments and become neurofibrillary tangles. This leads to malfunction of the structural and regulatory actions of the cytoskeleton and then leads to abnormal morphology, axonal transport, and synaptic function of neurons, thus leading to neurodegeneration [12].
These prior theories paved the way to the widely accepted hypothesis for the pathogenesis of AD: the amyloid cascade hypothesis. This theory attributes clinical sequelae of the disease to the overproduction or decreased clearance of amyloid beta (Ab) peptides, which then leads to increased deposition of Ab, furthermore, leading to neuronal damage (Figure 1). The length of Ab varies depending on the posttranslational cleavage pattern of the transmembrane amyloid precursor protein (APP). Ab is generated by cleavage of APP via either b- or g-secretases, resulting in the infamous insoluble Ab fibrils [13]. Two main types of Ab polymers play a direct role in the pathology of AD: Ab40 and Ab42. Ab40/Ab42 then oligomerizes, travels to synaptic clefts, and interferes with synaptic signaling. These eventually further polymerize into insoluble amyloid fibrils that aggregate into amyloid plaques [14]. Within the plaques, Ab peptides in b-sheet conformation polymerize into structurally distinct forms, including fibrillar, protofibers and polymorphic oligomers. It is the deposition of these plaques diffusely throughout the brain that lead to microglial activation, cytokine release, reactive astrocytosis, and an overall inflammatory response. These structural changes lead to synaptic and neuronal loss and eventual gross cerebral atrophy [12]. On the other hand, should APP be processed by a-secretase in the healthy adult, soluble b-amyloid is produced, which has been linked to play a role in neuronal plasticity/survival, is protective against excitotoxicity, is important for early CNS development, and has been shown to be important for promoting synapse formation [15].
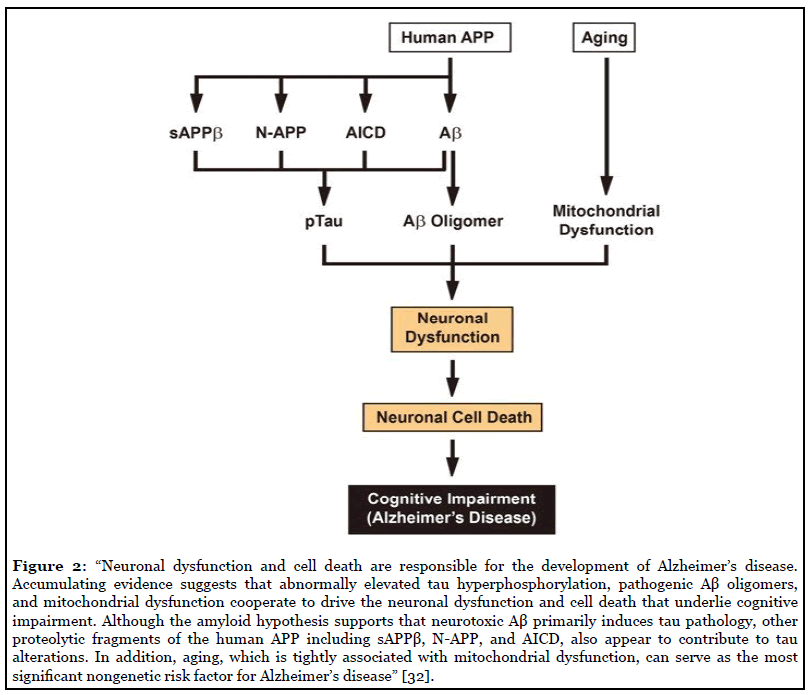
The genetics of AD should also be considered to play an influential role in the pathogenesis, alongside inflammation, apoptosis, and plaque buildup. In fact, the APP gene located on 21q21, mentioned above, was the first discovered causative gene of AD [13]. Advances in genetic research have identified two distinct forms of AD: Familial Alzheimer’s Disease (FAD) and Sporadic Alzheimer Disease (SAD), with the latter making up the majority of cases. Important advances in the 1990s and early 2000s revealed that FAD is the result of autosomal dominant mutations in APP, PSEN1 and PSEN2 genes, located on chromosome 21, 14, and 1, respectively [12,16]. More specifically, PSEN1 and PSEN2 contain the necessary amino acid residues required for the catalytic active site of gamma-secretase. Certain mutations of these genes lead to increased production of Ab peptide and neurodegeneration [12]. Far more commonly, the genetic risk factor for SAD was identified as the type e4 allele on chromosome 19, of the gene for apolipoprotein E (APOE), a low-density lipoprotein carrier resides. APOE is present in roughly 50- 60% of patients with AD compared to 20-25% in healthy elderly adults without the history of familial AD. APOE is associated with an approximately three-fold risk of developing AD if one copy is present, and there is an eightfold risk if two copies are present [16]. These plus other major influencing genes for SAD are listed in Table 1 and 2.
| Genes Involved in Pathogenesis of AD | Abbreviations |
|---|---|
| Amyloid Precursor Protein gene | APP |
| Presenilin gene | PS |
| Apolipoprotein E gene | APOE |
| Clusterin gene | n/a |
| Complement Receptor 1 gene | n/a |
| Phospholipids Bind to Clathrin Protein gene | PICALM |
| Cholesterol Metabolism gene | CH25H, ABCAL, and CH24H |
| Sterol O-acyltransferase gene | SOAT1 |
| Prostaglandin-endoperoxide Synthase 2 gene | Ptgs2 |
| Angiotensin-Converting Enzyme gene | n/a |
| SLC26A38 gene | n/a |
Table 1: A list of several genes that play a major influential role for the development of Sporadic Alzhemer’s Disease (SAD) [17].
| Gene | Proposed mechanism of dysfunction contributing to pathogenesis in Alzheimer’s disease |
| APP | Defects in synaptic development and neuronal migration [18], A-beta peptide synthesis defects [19] |
| PSEN 1 / 2 | Altered gamma-secretase activity resulting in elevated amyloid beta-42 levels [20] |
| APOE – epsilon 4 | Lipid and cholesterol dysmetabolism, synaptic inflammation, impaired clearance of amyloid beta-42, LDL receptor impairment [21-24] |
| CLU | Deposition and metabolism of amyloid beta-42 [25] |
| ABCA7 | Immune mediated and lipid metabolic response alterations [26] |
| CR1 | Aggravated senile plaque formation [27] |
| CD33 | Influences microglia mediated clearance of amyloid beta-42 [28] |
| MS4A | Dysregulation of intracellular calcium concentration [29] |
| EPHA1 | Axonal guidance changes [30] |
| SORL1, BIN, CD2AP, PICALM | Alterations in lipid metabolism [31] |
Table 2: A list of relevant genes involved in pathogenesis of Alzheimer’s disease.
Clinical Presentation
The most common presentation of AD is that of an elderly individual with an insidious progression of cognitive decline, most centered around memory loss. Declines in non-memory aspects of cognition including word-finding, vision/spatial issues, impaired reasoning or judgment, are also seen in early stages. At this time, patients meet criteria for mild cognitive impairment [33]. Almost all patients diagnosed with AD also have neuropsychiatric symptoms during some stage of their disease, of which depression and apathy are the most dominant early on. Verbal and physical aggression are frequently observed throughout all stages. As the disease progresses, delusions, hallucinations, and aggression are more often seen and additionally, circadian sleep-wake rhythms are more exaggerated as compared to those with normal aging [34]. As the disease progresses, cognitive difficulties become more apparent and widespread, eventually impacting activities of daily living. The decline in two or more areas, including memory, language, executive and visuospatial function, personality, and behavior that lead to the loss of the ability to perform instrumental and basic ADL’s point towards the diagnosis of AD dementia [33]. For those that do survive to the late stages of AD, death is often due to consequences of the disease itself as well as increased vulnerability to falls, pressure sores, and infections, ultimately leading to an average of 8 years from diagnosis to death [35]. Tools considered useful for clinical detection of AD include global cognitive screens, such as the MMSE and MOCA, and more specific tests of memory impairment like the five-word test. More formal diagnosis can be done by specialists, such as neuropsychologists [36].
FAD tends to have the typical presentation mentioned above, although at a much earlier age. Some PSEN1 mutations are associated with other features of disease, including seizures, spastic paresis, and myoclonus [33]. There are rarer forms of AD that present atypically, including posterior cortical atrophy, logopenic primary progressive aphasia, and frontal variant AD [35]. Posterior cortical atrophy tends to present with progressive loss of higher visual functions, such as diminished ability to interpret, locate, or reach for objects via visual guidance. A decreased ability of numeracy, literacy, and praxis may also be present in this variant of the disease [37]. Logopenic primary progressive aphasia presents more with language disturbance focus, characterized by slow word retrieval, word finding difficulties, and impaired sentence repetition, while motor speech, grammar, and single-word comprehension are spared [38]. The frontal variant of AD presents with stereotyped behaviors, progressive apathy/ behavior disinhibition, and executive dysfunction [39]. These variant presentations are important to address, but more than likely, they do result in the more global and typical picture of memory loss and dementia caused by AD over the course of time.
Histology
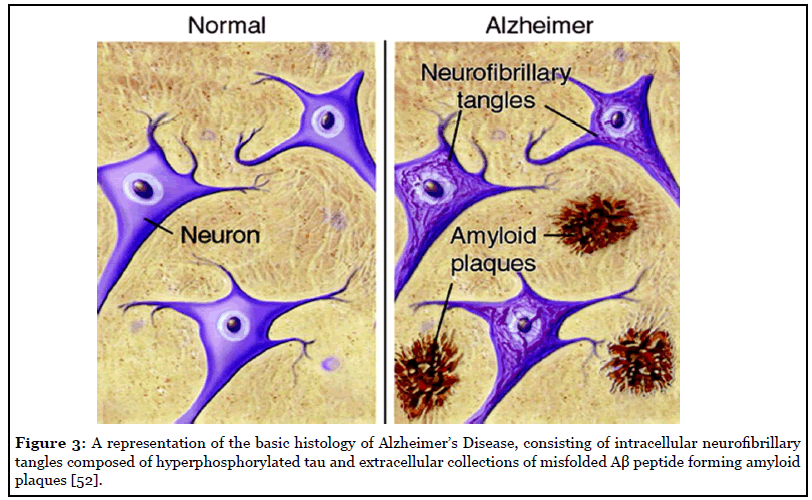
Amyloid plaques and neurofibrillary tangles are the cardinal features of Alzheimer histopathology. The amyloid plaques consist of extracellular accumulations of misfolded Ab sheets consisting of 40 or 42 amino acids (Ab40 and Ab42), the two by-products of amyloid precursor protein metabolism [33]. Amyloid plaques are located extracellularly, and initially develop in the basal, temporal, and orbitofrontal cortex of the brain, and later progress to involve the neocortex, hippocampus, amygdala, diencephalon, and basal ganglia. These aggregates of Ab trigger the formation of neurofibrillary tangles (NFS), which are composed mostly of hyperphosphorylated tau protein. These NFS are found in the locus coeruleus and transentorhinal and entorhinal areas of the brain. In the last critical stage, these histopathological entities are spread to the hippocampus and neocortex [14]. Additionally, neuropil threads, dystrophic neurites, associated astrogliosis, microglial activation and evidence of amyloid angiopathy are also common findings [33]. Histology images of Alzheimer’s Disease is illustrated in Figure 3.
Testing
AD remains a clinical diagnosis and is reliant on a detailed history, cognitive assessment and physical exam. Structural imaging is also an important component of the assessment for AD. Atrophy of the medial temporal lobes on magnetic resonance imaging (MRI) is considered a diagnostic marker for the mild cognitive impairment stage of AD [40]. Similarly, hypometabolism in the parieto-temporal association area, posterior cingulate and precuneus on fluorodeoxyglucose PET imaging is associated with AD [41].
In 2012, the Food and Drug Administration approved the first beta-amyloid tracer, florbetapir, for use in PET scans of suspected Alzheimer’s patients. In one study, florbetapir PET imaging was shown to have a 92% sensitivity and 100% specificity for detecting moderate to frequent plaques in patients with an autopsy within 2 years of the scan [42]. The amyloid PET tracers florbetaben and flutemetamol have also been approved and show similar sensitivity and specificity [43]. However, amyloid PET scans have had limited clinical impact due to lack of insurance reimbursement, and they are currently used primarily in research trials.
Biomarkers in CSF further support a diagnosis of AD. The biomarker profile of AD is increased total tau (T-tau) and phosphorylated tau (P-tau) and decreased amyloidbeta (Ab42). This biomarker pattern can also predict which subjects with MCI are likely to progress to AD [44]. Amyloid PET and the CSF marker Ab42 show a concordance of around 90% across several studies [45]. One longitudinal study of healthy elderly and patients with MCI showed similar diagnostic accuracy between CSF and amyloid PET biomarkers and no improvement when combining them [46]. Thus, the choice between CSF and amyloid PET biomarkers can be based on availability, cost, patient preference and other factors such as patient suitability for radiation or lumbar puncture.
Currently, there are no blood biomarkers routinely used to diagnose AD. Only a small fraction of brain proteins enters the bloodstream, and these proteins are diluted by plasma proteins such as albumin and IgG, making them difficult to measure quantitatively. Additionally, brain proteins in plasma may be degraded or metabolized, so that plasma levels would not reflect real-time changes in the brain [47]. Nonetheless, several plasma proteins are being examined as potential noninvasive biomarkers of AD. Plasma tau is one such protein. One study with prospective and crosssectional cohorts found higher plasma tau to be associated with AD, but with significant overlap in levels of healthy controls [48]. Another potential blood marker is the axonal neurofilament light (NFL) protein. A prospective study found high correlation between plasma NFL and CSF NFL, and high diagnostic accuracy of plasma NFL in patients with AD versus controls [49]. However, plasma NFL is also increased in other neurological diseases such as frontotemporal dementia, limiting its utility for differential diagnosis of AD [50]. Finally, despite the utility of CSF Ab42 as a biomarker, plasma Ab42 has not shown to be predictive of AD development in patients with MCI. Unlike NFL, there is a lack of correlation between levels of Ab42 in CSF and plasma, possibly due to the release of Ab42 in plasma from peripheral tissues [51].
Treatment
Currently, treatment for Alzheimer’s disease does not impact the progression or underlying pathology of the disease. However, with medication and other therapies, steps are taken to increase the quality of life of those with AD. Current therapeutics are based off the Cholinergic Theory, which attributes a decrease in cholinergic neurotransmission to a decline in cognitive function [12]. At present, there are two classes of pharmacologic therapy available for AD: cholinesterase inhibitors (donepezil, rivastigmine, and galantamine) and memantine, which has activity as both a non-competitive N-methyl-D-aspartate receptor antagonist and a dopamine agonist [3]. The cholinesterase inhibitors are approved for use in patients with mild, moderate, or severe AD dementia as well as Parkinson’s disease dementia. Memantine is approved for use in patients with moderate to severe AD who may also have difficulty with alertness and attention.
Other aims of treatment look to modifiable risk factors in one’s overall health and “cognitive reserve” including cardiovascular/lifestyle factors, such as a healthy diet and plenty of physical exercise, as well as cognitive engagement. Cognitive reserve refers to the ability to fend off pathologic insult, meaning the ability to engage alternate synaptic pathways or cognitive strategies to cope with the pathology of AD. By improving one’s physical wellbeing and mental reserve, this may delay clinical symptoms of AD [36]. Preventing, halting, or slowing down the progression of AD with nutrition has been a topic of much research in Alzheimer’s today. Antioxidants, omega-3 fatty acids, B vitamins, folate, medium chain triglycerides, and combination medical foods, are just a few of the avenues of which are being studied. For instance, the variable actions inherent in any given antioxidant, such as reducing oxidized membrane lipids, limiting damage to nucleic acids, and influencing strep kinase pathways, may alter the pathways of cellular injury that lead to the positive benefits of having them in the diet of someone with AD [53]. Research has also demonstrated Vitamin E to prevent AD-like changes in the brains of AD genetic models of mice, and other clinical trials have shown an increase in median survival in patients with AD who were treated with selegiline (15 mg twice daily) and α-tocopherol (1000 IU twice daily) [53]. Several large scale clinical trials have also looked into omega-3 fatty acids, one of which being the MIDAS study which found that 900 mg of daily docosahexaenoic acid (DHA) led to a 7 year age improvement in cognition over 24 weeks when compared to placebo. This study and many more point to DHA’s potential direct effects on neurodegeneration in AD as well as its overarching reduction in cerebrovascular disease [53]. To discuss DHA even further, a randomized control trial looked at the effect of omega-3 fatty acids had on B vitamin function. To preface, inadequate B vitamin status alone leads to the accumulation of homocysteine, a non-essential amino acid, that when elevated is recognized as a modifiable risk factor for AD and other dementias. This goes along with the results of the VITACOG trial, which showed that B vitamin supplement in elderly adults with mild cognitive impairment slowed the rate of brain atrophy both globally and regionally [54]. And so it makes sense that the group that was randomized to B vitamin treatment (folic acid and vitamins B6 and B12) showed that B vitamins plus an omega-3 level in the upper range of normal interacted to slow cognitive decline, basically sumizing that elevated omega-3 acids alone significantly enhanced the cognitive effect of B vitamins [54].
Novel treatments for AD today are based off multifaceted strategies and are currently under investigation via clinical trials. There are three trials currently ongoing, including gantenerumab, crenezumab, and aducanumab as immunotherapy approaches, which aim to effectively clear the Ab aggregates [55]. Other future targets for treatment include targeting mitochondrial dysfunction, targeting excitotoxicity and misfolding protein aggregations via novel acetylcholinesterase inhibitors or NMDA-Receptor Antagonists, targeting autophagy, and targeting neuroinflammation, to name a few [55]. Links have also been made regarding the gut microbiota and AD, and further research regarding dysbiosis and worsening disease are being investigated for other novel treatment approaches [56].
Future Directions
The majority of therapeutics in development target the two hallmark pathologies of AD: beta-amyloid plaques and neurofibrillary tangles of tau. As of February 2019, the pipeline of AD drugs includes 132 agents, with 96 (73%) intended to achieve disease modification. Of these, 38 have amyloid as a primary or combination target, and 17 have tau as a primary or combination target [57].
One class of therapeutics against amyloid, monoclonal antibodies, is intended to facilitate amyloid removal from the brain. Data have so far failed to show improvement in clinical symptoms despite significant reductions in amyloid load. However, one antibody which had previously failed futility analyses in Phase III trials later showed positive results in a subset of patients. The antibody, aducanumab, is directed against both amyloid fibrils and soluble oligomers, and its manufacturer Biogen plans to apply for FDA approval in 2020 [58]. Another set of therapeutics inhibit the enzyme that cleaves the beta-amyloid protein from its precursor, amyloid precursor protein (APP). These drugs against the beta-site APP-cleaving enzyme 1 (BACE-1) have similarly led to reductions in amyloid levels but produce worse cognitive decline in AD patients compared to placebo. The results indicate BACE-1 activity may be important in preserving normal synaptic function [59].
Several therapeutics are also in development to target tau, which is downstream of amyloid and thought to be the direct cause of AD symptoms. Although tau accumulates intracellularly, several in vivo and in vitro studies have shown tau can move from one neuron to another in a “prion-like” spread, suggesting a possible therapeutic target [60]. LMTX, a selective inhibitor of tau protein aggregation, failed to show cognitive improvement in AD patients compared to placebo in a Phase III trial, but a separate Phase II/III trial was started to test a lower dose of the drug [61]. Several anti-tau antibodies are in early clinical testing, as are at least two active tau vaccines designed to stimulate antibody production in AD patients [62].
Given the underwhelming results of therapeutics against amyloid and tau, drug development has shifted its focus to the “pre-dementia” space, including patients with preclinical AD and individuals with risk factors for cognitive decline. A large number of therapeutic trials with private and public funding are underway in high-risk asymptomatic individuals, including carriers of genetic mutations and those with amyloid-positive PET scans or elevated biomarkers [63]. Data from these trials in the next several years may provide insight into which interventions, if any, can slow or halt the onset of symptomatic AD.
Ethical Standards
This review article was written in a manner consistent with ethical and scientific guidelines regarding research and publication.
Contributions
Both Samantha McGirr and Courtney Venegas, fourth year medical students, were equally involved in the research and writing of this paper. Dr. Swaminathan, Neurologist at UNMC who contributed to and edited the published review, supervised them both.
References
2. Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia. 2020 Mar 10; 16(3):391-460.
3. Weller J, Budson A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research. 2018;7.
4. Alzheimer A. Uber eine eigenartige Erkrankung der Hirnrinde. Zentralbl. Nervenh. Psych. 1907;18:177-9.
5. Yang HD, Kim DH, Lee SB, Young LD. History of Alzheimer’s Disease. Dementia and Neurocognitive Disorders. 2016 Dec 1;15(4):115-21.
6. Soria Lopez JA, Gonzalez HM, and Leger GC. Chapter 13 – Alzheimer’s disease. Handbook of Clinical Neurology 2019; 167: 231-255.
7. Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochemical and Biophysical Research Communications. 1984 May 16;120(3):885-90.
8. Brion JP, Couck AM, Passareiro E, Flament-Durand J. Neurofibrillary tangles of Alzheimer’s disease: an immunohistochemical study. Journal of Submicroscopic Cytology. 1985 Jan 1;17(1):89-96.
9. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group* under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984 Jul 1;34(7):939-44.
10. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack Jr CR, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia. 2011 May;7(3):263-9.
11. Jack Jr CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimer’s & Dementia. 2018 Apr;14(4):535-62.
12. Barage SH, Sonawane KD. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides. 2015 Aug 1;52:1-8.
13. Shao W, Peng D, Wang X. Genetics of Alzheimer’s disease: From pathogenesis to clinical usage. Journal of Clinical Neuroscience. 2017 Nov 1;45:1-8.
14. Tiwari S, Atluri V, Kaushik A, Yndart A, Nair M. Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics. International Journal of Nanomedicine. 2019;14:5541.
15. Šerý O, Povová J, Míšek I, Pešák L, Janout V. Molecular mechanisms of neuropathological changes in Alzheimer’s disease: a review. Folia Neuropathologica. 2013;51(1):1-9.
16. Bondi MW, Edmonds EC, Salmon DP. Alzheimer’s disease: past, present, and future. Journal of the International Neuropsychological Society: JINS. 2017 Oct;23(9-10):818.
17. Chen YG. Research progress in the pathogenesis of Alzheimer’s disease. Chinese Medical Journal. 2018 Jul 5;131(13):1618.
18. Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J. Synapse formation and function is modulated by the amyloid precursor protein. Journal of Neuroscience. 2006 Jul 5;26(27):7212-21.
19. Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, et al. The’Arctic’APP mutation (E693G) causes Alzheimer’s disease by enhanced Aβ protofibril formation. Nature Neuroscience. 2001 Sep;4(9):887-93.
20. Schellenberg GD, Bird TD, Wijsman EM, Orr HT, Anderson L, Nemens E, et al.. Genetic linkage evidence for a familial Alzheimer’s disease locus on chromosome 14. Science. 1992 Oct 23;258(5082):668-71.
21. Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. Journal of Lipid Research. 2009 Apr 1;50(Supplement):S183-8.
22. Herz J, Chen Y, Masiulis I, Zhou L. Expanding functions of lipoprotein receptors. Journal of Lipid Research. 2009 Apr 1;50(Supplement):S287-92.
23. Zhong N, Weisgraber KH. Understanding the association of apolipoprotein E4 with Alzheimer disease: clues from its structure. Journal of Biological Chemistry. 2009 Mar 6;284(10):6027-31.
24. Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, et al. ApoE influences amyloid-? (A?) clearance despite minimal apoE/A? association in physiological conditions. Proceedings of the National Academy of Sciences. 2013 May 7;110(19):E1807-16.
25. Nuutinen T, Suuronen T, Kauppinen A, Salminen A. Clusterin: a forgotten player in Alzheimer’s disease. Brain Research Reviews. 2009 Oct 1;61(2):89-104.
26. Ramirez LM, Goukasian N, Porat S, Hwang KS, Eastman JA, Hurtz S, Wang B, Vang N, Sears R, Klein E, Coppola G. Common variants in ABCA7 and MS4A6A are associated with cortical and hippocampal atrophy. Neurobiology of Aging. 2016 Mar 1;39:82-9.
27. Chibnik LB, Shulman JM, Leurgans SE, Schneider JA, Wilson RS, Tran D, et al. CR1 is associated with amyloid plaque burden and age-related cognitive decline. Annals of Neurology. 2011 Mar;69(3):560-9.
28. Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, Rosenkrantz LL, Imboywa S, Lee M, Von Korff A, Morris MC. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nature Neuroscience. 2013 Jul;16(7):848-50.
29. Karch CM, Jeng AT, Nowotny P, Cady J, Cruchaga C, Goate AM. Expression of novel Alzheimer’s disease risk genes in control and Alzheimer’s disease brains. PloS one. 2012 Nov 30;7(11):e50976.
30. Wang HF, Tan L, Hao XK, Jiang T, Tan MS, Liu Y, Zhang DQ, Yu JT, Alzheimer’s Disease Neuroimaging Initiative. Effect of EPHA1 genetic variation on cerebrospinal fluid and neuroimaging biomarkers in healthy, mild cognitive impairment and Alzheimer’s disease cohorts. Journal of Alzheimer’s Disease. 2015 Jan 1;44(1):115-23.
31. Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nature Genetics. 2011 May;43(5):436-41.
32. Jeong S. Molecular and cellular basis of neurodegeneration in Alzheimer’s disease. Molecules and Cells. 2017 Sep 30;40(9):613.
33. Lane CA, Hardy J, Schott JM. Alzheimer’s disease. The European Journal of Neurology. 2018 Jan;25(1):59-70
34. Lyketsos CG, Carrillo MC, Ryan JM, Khachaturian AS, Trzepacz P, Amatniek J, et al. Neuropsychiatric Symptoms in Alzheimer’s Disease. Alzheimers & Dementia. 2011 Sep;7(5):532-9.
35. Eratne D, Loi SM, Farrand S, Kelso W, Velakoulis D, Looi JC. Alzheimer’s disease: clinical update on epidemiology, pathophysiology and diagnosis. Australasian Psychiatry. 2018 Aug;26(4):347-57.
36. Aisen PS, Cummings J, Jack CR, Morris JC, Sperling R, Frölich L, et al. On the path to 2025: understanding the Alzheimer’s disease continuum. Alzheimer’s Research & Therapy. 2017 Dec 1;9(1):60.
37. Crutch SJ, Schott JM, Rabinovici GD, Murray M, Snowden JS, van der Flier WM, et al. Consensus classification of posterior cortical atrophy. Alzheimer’s & Dementia. 2017 Aug;13(8):870-84.
38. Oh MJ, Kim S, Park YH, Suh J, Yi S. Early Onset Alzheimer’s Disease Presenting as Logopenic Primary Progressive Aphasia. Dementia and Neurocognitive Disorders. 2018 Jun 1;17(2):66-70.
39. Villain N, Dubois B. Alzheimer’s disease including focal presentations. InSeminars in neurology. Thieme Medical Publishers. 2019; p. 213-26.
40. Frisoni GB, Fox NC, Jack CR, Scheltens P, Thompson PM. The clinical use of structural MRI in Alzheimer disease. Nature Reviews Neurology. 2010 Feb;6(2):67-77.
41. Kato T, Inui Y, Nakamura A, Ito K. Brain fluorodeoxyglucose (FDG) PET in dementia. Ageing Research Reviews. 2016 Sep 1;30:73-84.
42. Clark CM, Pontecorvo MJ, Beach TG, Bedell BJ, Coleman RE, Doraiswamy PM, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-? plaques: a prospective cohort study. The Lancet Neurology. 2012 Aug 1;11(8):669-78.
43. Yeo JM, Waddell B, Khan Z, Pal S. A systematic review and meta-analysis of 18F-labeled amyloid imaging in Alzheimer’s disease. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring. 2015 Mar 1;1(1):5-13.
44. Hampel H, Bürger K, Teipel SJ, Bokde AL, Zetterberg H, Blennow K. Core candidate neurochemical and imaging biomarkers of Alzheimer’s disease. Alzheimer’s & Dementia. 2008 Jan 1;4(1):38-48.
45. Blennow K, Mattsson N, Schöll M, Hansson O, Zetterberg H. Amyloid biomarkers in Alzheimer’s disease. Trends in Pharmacological Sciences. 2015 May 1;36(5):297-309.
46. Palmqvist S, Zetterberg H, Mattsson N, Johansson P, Minthon L, Blennow K, et al, Alzheimer’s Disease Neuroimaging Initiative, Swedish BioFINDER Study Group. Detailed comparison of amyloid PET and CSF biomarkers for identifying early Alzheimer disease. Neurology. 2015 Oct 6;85(14):1240-9.
47. Blennow K, Zetterberg H. Biomarkers for Alzheimer’s disease: current status and prospects for the future. Journal of Internal Medicine. 2018 Dec;284(6):643-63.
48. Mattsson N, Zetterberg H, Janelidze S, Insel PS, Andreasson U, Stomrud E, et al. Plasma tau in Alzheimer disease. Neurology. 2016 Oct 25;87(17):1827-35.
49. Mattsson N, Andreasson U, Zetterberg H, Blennow K. Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurology. 2017 May 1;74(5):557-66.
50. Rohrer JD, Woollacott IO, Dick KM, Brotherhood E, Gordon E, Fellows A, et al. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology. 2016 Sep 27;87(13):1329-36.
51. Hansson O, Zetterberg H, Vanmechelen E, Vanderstichele H, Andreasson U, Londos E, et al. Evaluation of plasma Aβ40 and Aβ42 as predictors of conversion to Alzheimer’s disease in patients with mild cognitive impairment. Neurobiology of Aging. 2010 Mar 1;31(3):357-67.
52. Silbert LC. Does statin use decrease the amount of Alzheimer disease pathology in the brain?. Neurology. 2007 Aug 28;69(9):E8-11.
53. Swaminathan A, Jicha GA. Nutrition and prevention of Alzheimer’s dementia. Frontiers in Aging Neuroscience. 2014 Oct 20;6:282.
54. Oulhaj A, Jernerén F, Refsum H, Smith AD, de Jager CA. Omega-3 fatty acid status enhances the prevention of cognitive decline by B vitamins in mild cognitive impairment. Journal of Alzheimer’s Disease. 2016 Jan 1;50(2):547-57.
55. Van Bulck M, Sierra-Magro A, Alarcon-Gil J, Perez- Castillo A, Morales-Garcia JA. Novel approaches for the treatment of Alzheimer’s and Parkinson’s disease. International Journal of Molecular Sciences. 2019 Jan;20(3):719.
56. Jiang C, Li G, Huang P, Liu Z, Zhao B. The gut microbiota and Alzheimer’s disease. Journal of Alzheimer’s Disease. 2017 Jan 1;58(1):1-5.
57. Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer’s disease drug development pipeline: 2019. Alzheimer’s & Dementia: Translational Research & Clinical Interventions. 2019 Jan 1;5:272-93.
58. Schneider L. A resurrection of aducanumab for Alzheimer’s disease. The Lancet Neurology. 2020 Feb 1;19(2):111-2.
59. Knopman DS. Lowering of amyloid-beta by β-secretase inhibitors—some informative failures. The New England Journal of Medicine. 2019 Apr 11;380(15):1476-1478
60. DeVos SL, Corjuc BT, Oakley DH, Nobuhara CK, Bannon RN, Chase A, et al. Synaptic tau seeding precedes tau pathology in human Alzheimer’s disease brain. Frontiers in Neuroscience. 2018 Apr 24;12:267.
61. Gauthier S, Feldman HH, Schneider LS, Wilcock GK, Frisoni GB, Hardlund JH, et al. Efficacy and safety of tauaggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. The Lancet. 2016 Dec 10;388(10062):2873-84.
62. Gauthier S, Feldman HH, Schneider LS, Wilcock GK, Frisoni GB, Hardlund JH, et al. Efficacy and safety of tauaggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. The Lancet. 2016 Dec 10;388(10062):2873-84.
63. Aisen P, Touchon J, Amariglio R, Andrieu S, Bateman R, Breitner J, et al. EU/US/CTAD task force: lessons learned from recent and current Alzheimer’s prevention trials. The Journal of Prevention of Alzheimer’s Disease. 2017;4(2):116.