Abstract
Diffuse cystic lung diseases are infrequently encountered in pathology practice, yet when present, often pose significant diagnostic challenges. They typically follow unclassical or atypical radiological findings, requiring pathology for a definite diagnosis and appropriate management. Another common scenario involves specimens obtained following surgery for spontaneous pneumothorax, which are submitted as part of routine postoperative evaluation. The accurate diagnosis of cystic lung disease is critical, as it directly impacts patient management. While radiologic imaging often serves as the initial guide in identifying cystic lung lesions, histopathologic correlation is essential for definitive diagnosis. This article is centered on histology and aims to understand the histologic hallmarks and identification of some of these cystic lung entities, with the goal of helping pathologists to arrive at a confident and clinically meaningful diagnosis.
Keywords
Cystic lung disease, Histopathology, Intraparenchymal cysts, Lymphangioleiomyomatosis, Birt-Hogg-Dubé syndrome, Pulmonary Langerhans cell histiocytosis, Lymphocytic interstitial pneumonia, Placental transmogrification, Differential diagnosis
Introduction
The mechanisms underlying pulmonary cyst formation involve remodeling of the lung parenchyma through displacement, destruction, or replacement of normal pulmonary architecture. The pathophysiologic processes contributing to this remodeling commonly include inflammation, infection, interstitial lung diseases, smoking-related changes, and genetic disorders [1]. These mechanisms collectively contribute to the spectrum of morphologic patterns seen in cystic lung diseases, which can be subtle pathologically and represent a diagnostic challenge for practicing pathologist.
Pathologically, pulmonary cysts are defined as discrete air-filled space lined by epithelium or surrounded by a thin fibrous wall (<2 mm). Diffuse cystic lung diseases (DCLDs) correspond to the “multifocal/diffuse cysts” category and represent a heterogeneous group of entities characterized radiologically by multiple thin-walled cysts within the lung parenchyma. High-resolution computed tomography (HRCT) can often distinguish true cysts from other air-containing lesions, including cavities (thick, irregular walls, often >4 mm), bullae (>1 cm with imperceptible walls), and bronchiectasis (dilated airways) [2–4].
As outlined in Figure 1, cystic lung diseases can be broadly categorized into solitary/incidental cysts and multifocal/diffuse cysts. Solitary/incidental cysts typically present as one or a few localized lesions and include incidental cysts, bronchogenic cysts, and pneumatoceles. These lesions are often identified incidentally and are generally straightforward to classify when interpreted in the appropriate clinical and radiologic context. For example, incidental cysts are commonly seen in older individuals, whereas intrapulmonary bronchogenic cysts are typically diagnosed in children or young adults, although they may also be detected incidentally in asymptomatic adults. Pneumatoceles are acquired, thin-walled cystic spaces that arise in the setting of acute infection or trauma and are typically self-limited.
Multifocal/diffuse cysts can be broadly divided into common and uncommon entities, as illustrated in Figure 1. Although common entities are frequently encountered in pathology practice, cystic changes are typically secondary phenomena associated with other dominant radiologic and histologic features. Common causes of cystic lung disease include smoking-related emphysema, usual interstitial pneumonia (UIP) with honeycombing and traction bronchiectasis, and cystic parenchymal remodeling following infection or malignancy. These entities are often accompanied by additional HRCT and histopathologic clues. Smoking-related cystic changes typically show a subpleural or paraseptal distribution. Histologically, they demonstrate intra-alveolar accumulation of finely pigmented macrophages, with or without associated smoking-related interstitial fibrosis. This fibrosis is characterized by a relatively uniform, dense, hypocellular interstitial fibrosis predominantly involving subpleural and paraseptal regions. In UIP and other fibrotic interstitial lung diseases, honeycombing and traction bronchiectasis correspond to well-established interstitial fibrosis on both radiologic and histologic evaluation. Cystic changes secondary to infection or malignancy-related remodeling are usually accompanied by additional diagnostic features, such as necrotizing inflammation, organizing pneumonia, or dominant nodules and masses, depending on the underlying process.
In contrast, a subset of uncommon diffuse lung diseases demonstrates more distinctive clinicopathologic features and often require a higher index of diagnostic suspicion. Although rare, these entities may show subtle but characteristic histopathologic findings that, when integrated with radiologic features, facilitate diagnosis. Uncommon entities include Birt-Hogg-Dubé (BHD) syndrome, lymphoid interstitial pneumonia (LIP), lymphangioleiomyomatosis (LAM), pulmonary Langerhans cell histiocytosis (PLCH), and placental transmogrification. While these entities may demonstrate characteristic imaging patterns, overlap is not uncommon in clinical practice. Findings such as ground-glass opacities and nodular lesions may be seen across multiple entities, and many conditions exhibit overlapping or subtle histologic features, further increasing diagnostic complexity. This review focuses on the histologic features of uncommon entities within the multifocal/diffuse cyst category encountered in routine pathology practice.
Birt-Hogg- Dubé (BHD) Syndrome
Etiology/underlying genetic abnormality
BHD syndrome is a rare autosomal dominant condition caused by germline mutations in the FLCN (folliculin) gene, with an estimated prevalence of approximately 1 in 200,000 individuals.
Clinical manifestations
The syndrome is characterized by an increased risk of cutaneous lesions (including fibrofolliculomas, trichodiscomas, and acrochordons), cystic lung disease, and renal neoplasms [5]. Fibrofolliculomas, known as benign cutaneous hamartomas, represent the most characteristic and often earliest manifestation of BHD syndrome and typically present within the age range of 20–40 years old. Pulmonary involvement is also another major feature of the disorder. Multiple bilateral pulmonary cysts occur in a substantial proportion of affected individuals and, in some cases, may represent the primary or sole clinical manifestation. Recurrent pneumothoraces are also common, with a peak incidence in the third to fifth decades of life. Renal neoplasms represent the most clinically significant systemic complication of BHD syndrome, occurring in approximately 12–34% of patients. These tumors typically occur around the age of 50, though they have been reported in patients ranging from 30 to 70 years of age [8].
Radiologic presentation
BHD-associated cysts are typically multiple, thin-walled, and sharply defined, with a predilection for the lower lobes and frequent localization along interlobular septa or bronchovascular bundles. Distinctive imaging features such as intra-cystic septa and protruding venules help differentiate BHD from other cystic lung diseases [6].
Histopathology
Pulmonary cysts in BHD are lined by alveolar epithelium and usually lack significant inflammation or fibrosis. They commonly abut interlobular septa (~88%), with intracystic septa (~14%) and protruding venules (~40%) occasionally identified on histopathologic examination [7].
Representative case
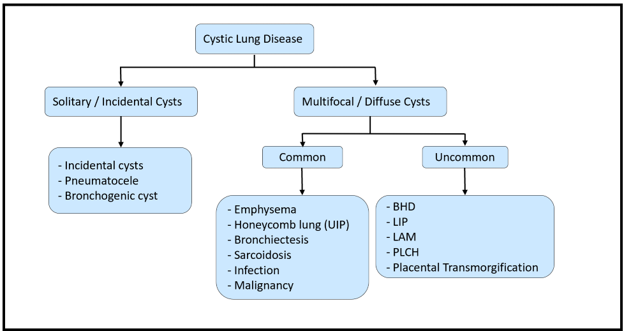
A representative case highlights the classic pulmonary manifestations of BHD syndrome. A 57-year-old female with a smoking history and prior spontaneous pneumothorax presented with recurrent right-sided pneumothorax following recent surgery. As shown in Figure 2, CT imaging revealed numerous thin-walled cysts involving all lobes of the lung, with distribution in the subpleural, peri-fissural, and paramediastinal regions. The cysts displayed oblong contours and variable sizes, with one measuring up to 4.9 × 2.1 cm. Histologic examination of wedge resections from the right lower lobe revealed numerous blebs with associated chronic fibrinous pleuritis. Multiple variably sized pulmonary cysts were surrounded by otherwise unremarkable lung parenchyma, without significant inflammation or fibrosis. While these histologic findings are non-specific, there were no features to suggest other well-defined cystic lung diseases. Subsequent review of the clinical history confirmed a remote diagnosis of BHD syndrome. Although subtle and potentially overlooked in limited tissue samples, these histologic features should prompt further workup to rule out BHD-related cystic disease. Recognition of this entity based on such findings is crucial and may have significant implications for both the patient and their family.
Lymphocytic Interstitial Pneumonia (LIP)
Etiology/underlying genetic abnormality
LIP is a rare benign lymphoproliferative interstitial lung disease that occurs most frequently in middle-aged adults, with a female-to-male ratio exceeding 2:1. It is strongly associated with systemic autoimmune disorders and immunodeficiency states [9,10]. LIP also represents one of the most common pulmonary complications in pediatric HIV infection, affecting up to 50% of HIV-positive children.
Representative case (with radiologic findings and histopathology)
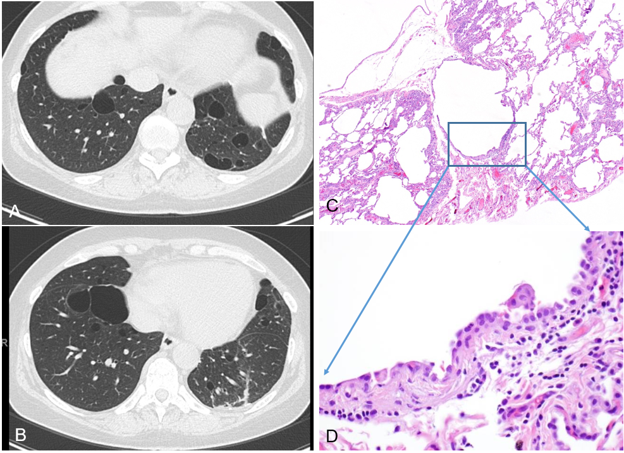
As shown in Figure 3, a 49-year-old man with a known history of Sjögren syndrome presented with exertional dyspnea. Pulmonary function testing was otherwise normal and an alpha-1 antitrypsin level was within normal limits. Radiologic imaging demonstrated diffuse ground-glass opacities, centrilobular nodules, and interlobular septal thickening, accompanied by scattered thin-walled cysts without significant fibrosis. Histologic evaluation of a right upper lobe wedge resection demonstrated a dense lymphoplasmacytic infiltrate expanding the alveolar septa, with peribronchiolar and perivascular lymphoid aggregations, and focal intra-alveolar proteinaceous exudates: findings characteristic of LIP. No lymphoepithelial lesions or clonal/atypical lymphoid populations were identified. Importantly, LIP may overlap with or progress to low-grade B-cell lymphoma, most commonly extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT). This progression is particularly associated with underlying systemic autoimmune disorders. Therefore, lung biopsy in suspected LIP or other atypical interstitial lung diseases is performed not only to establish a specific diagnosis but also to exclude an underlying lymphoproliferative neoplasm.
Lymphangioleiomyomatosis (LAM)
Etiology/underlying genetic abnormality
LAM is a rare cystic lung disease that predominantly affects women and is characterized by diffuse cystic destruction of the lung parenchyma [11]. The disease occurs either sporadically or in association with tuberous sclerosis complex (TSC), with the sporadic form accounting for the majority of cases due to somatic TSC2 mutations [12].
Clinical manifestations
LAM most often presents with progressive dyspnea and recurrent pneumothoraces, which occur in a substantial proportion of patients, and may also be accompanied by hemoptysis or chylous pleural effusions [11].
Radiologic presentation
Radiographically and grossly, the lungs demonstrate numerous thin-walled cysts distributed diffusely throughout the parenchyma, predisposing to air trapping and spontaneous rupture.
Histopathology
Histologically, LAM is defined by cystic airspaces lined or surrounded by groups of spindle-shaped smooth muscle-like cells (LAM cells). These cells are characterized by bland spindle, oval-to-fusiform nuclei and abundant eosinophilic cytoplasm without significant atypia or clear cell change. LAM cells may be subtle and are often associated with hemorrhage and hemosiderin-laden macrophages, posing diagnostic challenges when rare tumor cells are obscured within an extensive hemorrhagic or inflammatory background. Immunohistochemically, they are positive for HMB-45, smooth muscle actin, and estrogen/progesterone receptors, while lacking expression of cytokeratin and S100. Pathogenetically, dysregulated mTOR signaling drives disease progression. Treatment with mTOR inhibitors such as sirolimus has been shown to stabilize lung function and improve clinical outcomes [13], while lung transplantation remains a consideration in advanced refractory disease. Timely and accurate diagnosis of LAM is essential for appropriate management.
Representative case
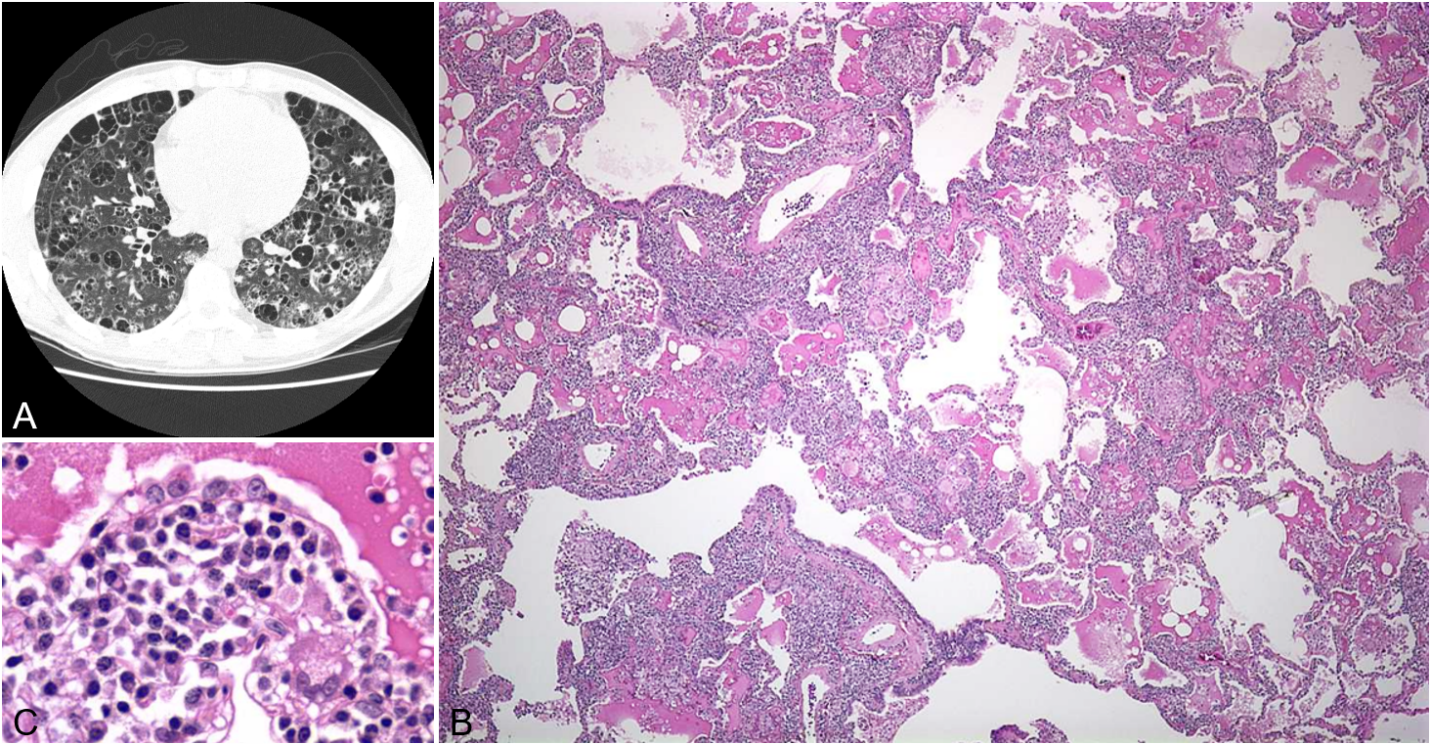
A 45-year-old woman presented with spontaneous left pneumothorax and underwent pleurodesis for persistent air leak. Chest CT revealed multiple, round, small, thin-walled cysts scattered diffusely throughout both lungs, with background ground-glass opacities (Figure 4A). Histologic sampling from a wedge resection showed diffuse chronic interstitial pneumonia with multiple cystic airspaces. On close examination, rare scattered aggregations of bland spindle cells with oval to fusiform nuclei and eosinophilic cytoplasm were identified along the cyst lining, admixed with numerous chronic inflammatory cells (Figure 4C). In addition, aggregates of bland clear cells lining lymphatic channels were found, easily overlooked on initial examination (Figure 4B). Immunohistochemical staining showed that these cells were positive for HMB-45, confirming the diagnosis of LAM. In this case, the histologic findings were subtle due to the extensive background inflammation and the sparse distribution of tumor cells. This highlights the importance of routinely considering and performing HMB-45 immunostaining in women with diffuse cystic lung disease to avoid missing the diagnosis when histologic features are inconspicuous.
Pulmonary Langerhans Cell Histiocytosis (PLCH)
Etiology/underlying genetic abnormality
PLCH is a rare, smoking-related cystic lung disease in adults that may occur in isolation or in association with involvement of other organ systems. It demonstrates a strong association with cigarette smoking, with more than 90% of patients being current or former smokers.
Clinical manifestations
PLCH may be complicated by recurrent pneumothoraces or progressive respiratory insufficiency in a subset of patients [14].
Radiologic presentation
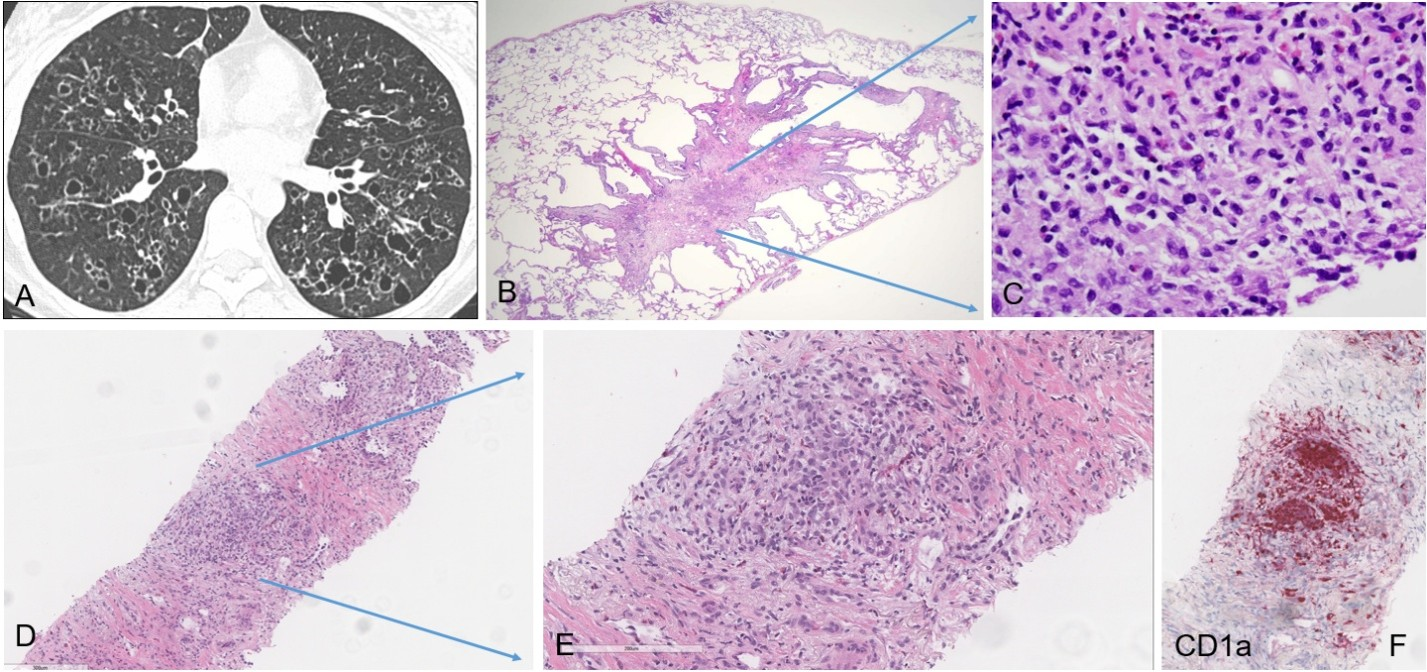
Radiographically, PLCH commonly exhibits an upper lobe predominant distribution of irregular nodules and cystic spaces (Figure 5A), which may coalesce in advanced disease, resulting in fibrotic scarring of variable size. HRCT typically reveals a characteristic combination of centrilobular nodules and irregularly shaped cysts with upper and mid-lung predominance as well as relative sparing of the costophrenic angles [15].
Histopathology
PLCH is characterized by nodular or stellate lesions composed of Langerhans cells admixed with variable numbers of eosinophils and other chronic inflammatory cells, set within a background of fibrotic or emphysematous lung parenchyma (Figure 5B). The Langerhans cells are haphazardly arranged and exhibit an irregular membrane, fine chromatin, and characteristic nuclear grooves (Figure 5C). They are immunoreactive for CD1a, S100, and Langerin (CD207), with Birbeck granules on electron microscopy confirming their lineage. Molecular studies have demonstrated activating mutations in MAPK pathway genes, including BRAF and MAP2K1 in a substantial subset of cases, supporting the concept of PLCH as a clonal neoplastic process rather than a purely reactive disorder [16].
Representative case
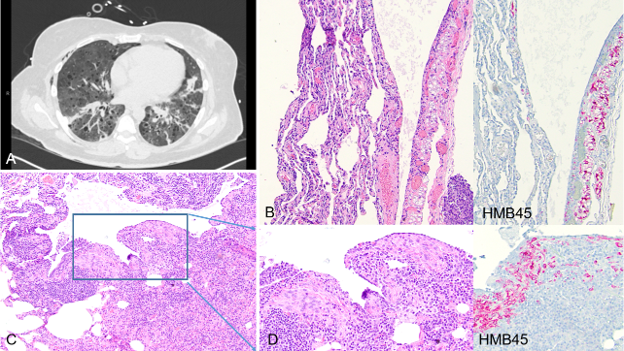
PLCH may present as a solitary nodular lesion mimicking pulmonary carcinoma, posing a diagnostic challenge [17,18]. Classic histomorphologic features may not be representative in smaller biopsy specimens. Therefore, recognition of key morphologic elements is critical to guide appropriate, targeted immunohistochemical workup, particularly in the current era of tissue preservation in limited lung biopsies. A 68-year-old female with a 20 pack-year smoking history presented with a new spiculated pulmonary nodule and interval enlargement of a previously identified RUL nodule, now measuring 12 × 9 mm (previously 8 × 7 mm), with a mildly increased SUV of 3.1, raising concern for malignancy. FNA of the RUL nodule demonstrated lung parenchyma with a consolidated area composed of haphazardly arranged epithelioid cells admixed with inflammatory cells, including eosinophils, in a background of dense fibrotic and fibroelastotic stroma (Figure 5D). No significant cytologic atypia or definitive malignant architecture was identified (Figure 5E). Immunohistochemical studies performed on this limited biopsy showed strong positivity for S100 and CD1a in aggregates of epithelioid cells (Figure 5F), supporting a diagnosis of PLCH, in the appropriate clinical context of a smoking history and eosinophil-rich inflammatory background. This case highlights that subtle histomorphologic clues in the appropriate clinical context, particularly a smoking history, combined with targeted immunohistochemistry are critical for establishing the correct diagnosis.
Placental Transmogrification of the Lung (PTL)
Etiology/underlying genetic abnormality
PTL is a rare and distinctive histologic entity associated with cystic lung disease, often occurring in the setting of smoking, and was first described in 1979 [19].
Clinical manifestations
PTL most commonly presents as unilateral bullous disease or, less frequently, as a solitary pulmonary nodule, and is often identified incidentally during evaluation of dyspnea, chest pain, recurrent pneumothoraces, or chronic obstructive pulmonary disease [20].
Radiologic presentation
On imaging, PTL typically appears as a large unilateral cystic or bullous lesion that may closely mimic emphysematous change or pulmonary neoplasm [21,22].
Histopathology
The hallmark feature is the presence of villus-like papillary projections resembling immature chorionic villi but lacking functional placental elements. These projections are lined by benign pneumocytes (flat, cuboidal, or occasionally ciliated) and contain fibrovascular cores composed of proliferating blood vessels, inflammatory cells, and adipose tissue, with occasional hyalinizing changes [23]. Immunohistochemical studies commonly demonstrate TTF-1 positivity in the epithelial lining, supporting a pulmonary epithelial origin [23]. Two principal pathogenetic theories have been proposed: one suggests that PTL represents localized alveolar destruction associated with emphysematous bullae, while the other favors a benign interstitial cell proliferation with cystic remodeling, analogous to pulmonary hamartomas or lipomatosis. Surgical resection is both diagnostic and curative, as PTL behaves in a benign fashion with no reported risk of recurrence or malignant transformation [24].
Representative case
A 63-year-old male with a history of heavy smoking presented with a radiologic impression of a ~1 cm solid pulmonary nodule on CT and PET/CT, suspicious for malignancy (Figure 6A). However, no malignancy was identified upon thorough examination of the wedge resection specimen. Instead, multiple variably sized villus-like papillary projections were present on histology, lined by bland pneumocytes and composed of proliferating blood vessels, inflammatory cells, and loose to hyalinizing stroma (Figure 6B, 6C). This distinctive histology may account for the radiological appearance of a solid nodule although it may not be fully appreciated histologically due to tangential sectioning. This discrepancy underscores the importance of recognizing the histologic features of PTL, which may present radiologically as a solid or cystic lesion and can mimic neoplasm.
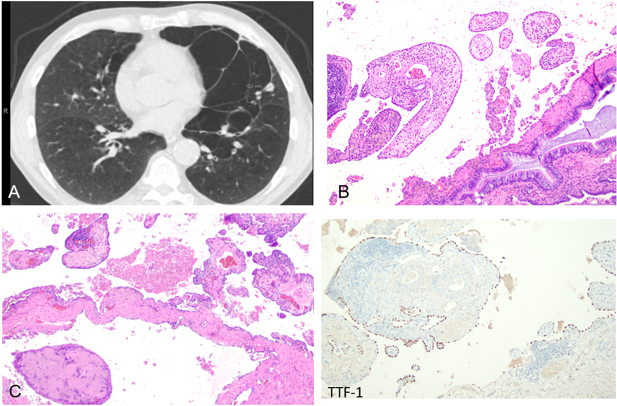
Figure 6. Placental Transmogrification of the Lung (PTL) Presenting as a Pulmonary Nodule. (A) CT imaging shows a solid nodule measuring approximately 1 cm in the left upper lobe, with mildly increased uptake on PET/CT. (B) Histologic examination reveals villous-like papillary structures composed of proliferating blood vessels, inflammatory cells, and loose to hyalinizing stroma (C). The lining epithelial cells are highlighted by TTF-1 immunohistochemical staining.
Discussion
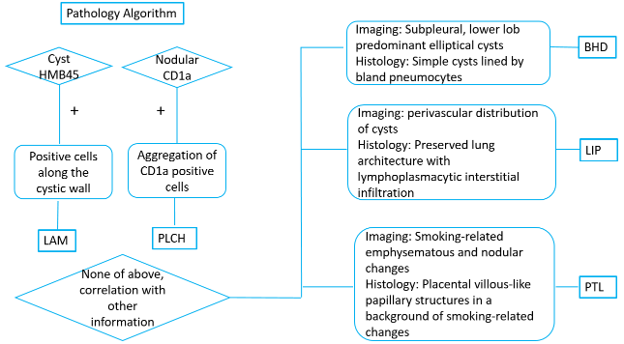
This review proposes a pathology-centered, stepwise diagnostic framework for evaluating intraparenchymal lung cysts, integrating radiologic classification with histologic criteria, particularly in cases with non-classic radiologic presentations encountered during pathologic evaluation. We highlight several uncommon diffuse cystic lung diseases that have not been thoroughly summarized in the literature and may be underrecognized in practice, especially in the absence of classic clinical presentations. For each entity, including BHD, LIP, LAM, PLCH, and PTL, we emphasize key diagnostic clues and practical workflows for accurate classification. In most cases, integration of clinical history and radiologic findings facilitates these diagnoses. However, atypical histologic or radiologic presentations, particularly in LAM, PLCH, and PTL, pose significant diagnostic challenges. In such cases, immunohistochemical stains including HMB45 and CD1a are recommended to avoid missing diagnoses when histologic features are non-classic (Figure 7). Smoking-related lung disease commonly demonstrates cystic changes, including emphysema, PLCH, and PTL. In this setting, associated nodular lesions may raise concern for malignancy due to increased PET uptake, irregular borders, and interval changes in size or morphology on imaging follow-up. When evaluating cystic lung lesions, pathologists must recognize these uncommon scenarios and integrate clinical, radiologic, and histologic findings. In such cases, the pathologist plays a central role in guiding accurate diagnosis and, consequently, appropriate clinical management.
References
2. Raoof S, Bondalapati P, Vydyula R, Ryu JH, Gupta N, Raoof S, et al. Cystic Lung Diseases: Algorithmic Approach. Chest. 2016 Oct;150(4):945–65.
3. Lee KC, Kang EY, Yong HS, Kim C, Lee KY, Hwang SH, et al. A Stepwise Diagnostic Approach to Cystic Lung Diseases for Radiologists. Korean J Radiol. 2019 Sep;20(9):1368–80.
4. Franciosi AN, Gupta N, Murphy DJ, Wikenheiser-Brokamp KA, McCarthy C. Diffuse Cystic Lung Disease: A Clinical Guide to Recognition and Management. Chest. 2025 Feb;167(2):529–47.
5. Menko FH, van Steensel MA, Giraud S, Friis-Hansen L, Richard S, Ungari S, et al. Birt-Hogg-Dubé syndrome: diagnosis and management. Lancet Oncol. 2009 Dec;10(12):1199–206.
6. Choi YJ, Park CH, Park HJ, Shin JM, Kim TH, Lee KA, et al. Characteristic Chest Computed Tomography Findings for Birt-Hogg-Dube Syndrome Indicating Requirement for Genetic Evaluation. Diagnostics (Basel). 2023 Jan 5;13(2):198.
7. Kumasaka T, Hayashi T, Mitani K, Kataoka H, Kikkawa M, Tobino K, et al. Characterization of pulmonary cysts in Birt-Hogg-Dubé syndrome: histopathological and morphometric analysis of 229 pulmonary cysts from 50 unrelated patients. Histopathology. 2014 Jul;65(1):100–10.
8. Pavlovich CP, Grubb RL 3rd, Hurley K, Glenn GM, Toro J, Schmidt LS, et al. Evaluation and management of renal tumors in the Birt-Hogg-Dubé syndrome. J Urol. 2005 May;173(5):1482–6.
9. Panchabhai TS, Farver C, Highland KB. Lymphocytic Interstitial Pneumonia. Clin Chest Med. 2016 Sep;37(3):463–74.
10. Koss MN, Hochholzer L, Langloss JM, Wehunt WD, Lazarus AA. Lymphoid interstitial pneumonia: clinicopathological and immunopathological findings in 18 cases. Pathology. 1987 Apr;19(2):178–85.
11. McCormack FX. Lymphangioleiomyomatosis: a clinical update. Chest. 2008 Feb;133(2):507–16.
12. Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci U S A. 2000 May 23;97(11):6085–90.
13. Taveira-DaSilva AM, Moss J. Clinical features, epidemiology, and therapy of lymphangioleiomyomatosis. Clin Epidemiol. 2015 Apr 7; 7:249–57.
14. Vassallo R, Ryu JH, Colby TV, Hartman T, Limper AH. Pulmonary Langerhans'-cell histiocytosis. N Engl J Med. 2000 Jun 29;342(26):1969–78.
15. Brauner MW, Grenier P, Tijani K, Battesti JP, Valeyre D. Pulmonary Langerhans cell histiocytosis: evolution of lesions on CT scans. Radiology. 1997 Aug;204(2):497–502.
16. Roden AC, Hu X, Kip S, Parrilla Castellar ER, Rumilla KM, Vrana JA, et al. BRAF V600E expression in Langerhans cell histiocytosis: clinical and immunohistochemical study on 25 pulmonary and 54 extrapulmonary cases. Am J Surg Pathol. 2014 Apr;38(4):548–51.
17. Song H, Lee D, Park SY, An YS, Yoon JK, Lee SJ. Single Pulmonary Langerhans Cell Histiocytosis Mimicking Metastasis in Lung Cancer: Imaging with (18)F-FDG PET/CT. Nucl Med Mol Imaging. 2015 Sep;49(3):243–5.
18. Suri HS, Yi ES, Nowakowski GS, Vassallo R. Pulmonary langerhans cell histiocytosis. Orphanet J Rare Dis. 2012 Mar 19; 7:16.
19. McChesney T. Placental transmogrification of the lung: a unique case with remarkable histopathologic features. Lab Investig 1979; 40: 245–6.
20. Ma DJ, Liu HS, Li SQ, Zhou XY, Cui YS, Wu HW, et al. Placental transmogrification of the lung: Case report and systematic review of the literature. Medicine (Baltimore). 2017 Sep;96(35):e7733.
21. Ferretti GR, Kocier M, Moro-Sibilot D, Brichon PY, Lantuejoul S. Placental transmogrification of the lung: CT-pathologic correlation of a rare pulmonary nodule. AJR Am J Roentgenol. 2004 Jul;183(1):99–101.
22. Kim JW, Park IH, Kwon W, Eom MS, Kim YJ, Oh JH. Placental transmogrification of the lung. Korean J Radiol. 2013 Nov-Dec;14(6):977–80.
23. Cavazza A, Lantuejoul S, Sartori G, Bigiani N, Maiorana A, Pasquinelli G, et al. Placental transmogrification of the lung: clinicopathologic, immunohistochemical and molecular study of two cases, with particular emphasis on the interstitial clear cells. Hum Pathol. 2004 Apr;35(4):517–21.
24. Zhou HY, Li H, Lu YJ, Wu HP, Li YZ. Placental transmogrification of the lung: two case reports and a literature review. Front Med (Lausanne). 2025 Sep 2; 12:1620403.