Abstract
Movement disorders may vary with the time of day and with mental state. Despite hypothalamic control of biological timing, we discuss how many cell populations throughout the brain and body, including dopaminergic nuclei, likely have time-keeping functions. We provide a “primer” of movement disorders with significant variation in time or with presentation in a state of altered consciousness. Parkinsonism illustrates our overall argument. Patients with an idiopathic behavioral and motoric disorder in sleep develop a parkinsonism eventually, at a high phenotype conversion rate over roughly ten years. Diurnal variation in severity of motor manifestations happens after diagnosis of a parkinsonism, and nocturnal movements of various types also occur after diagnosis. An inflammatory or autoimmune mechanism for timing or state variability in movement disorders may be part of the pathophysiology in many Parkinsonisms, but autoimmunity as a disease mechanism becomes particularly relevant in various encephalitides associated with movement disorders. We address seven major syndromes. Further addressing temporal variability, we then discuss Willis-Ekbom (restless legs) syndrome, an akathisia that implicates both iron and dopamine. Clinically, it is a multi-faceted movement disorder associated with either recumbency or with sleep. Next, we discuss dystonias characterized specifically by variation during the day. The clinical implications of our review are several: Parkinsonism is not exclusively a motor disease; identification of movement disorders is increasingly important in encephalitic conditions; state-dependency (in altered consciousness, in repose, in sleep) and temporal variation are hallmarks of many dyskinesias. We end with observations about the relevance of time in the assessment of disordered movement.
Keywords
Diurnal, Circadian, Parkinsonism, Synucleinopathy, Surface and cytosolic antibodies, Restless legs, Dopamine, Monogenic dystonia
Introduction
Clinicians are familiar with a question they should routinely ask of patients: are there factors that elicit or exacerbate a problem? In movement disorders, a relevant question might be: is there a time of day when a motor problem (a dyskinesia, such as restlessness or akathisia) occurs especially? It is a truism that many movement disorders, with the exception perhaps of myoclonus, lessen or dissipate during sleep [1]. Yet dyskinesias may manifest at different times in a day, in sleep, or in a state of altered consciousness. We examine the variety of conditions in which time-of-day or state-associated motor manifestations matter in reaching a correct diagnosis.
Our interest in movement disorders in the time domain stems from past studies, reviewed in [2], that explore how planned and spontaneous movements and transitions in behavior may involve dopamine neurons (DANs) as time keepers. In animal studies, DAN firing modulates a propensity to movement; the firing does not trigger movement per se [2]. In humans, the appearance of adventitious movements related to time of day, to wakefulness or sleep, or to an altered state of consciousness invites consideration of a role that dopamine (DA) or other mechanisms might play in generating such time-associated propensity to movement or dyskinesia. Disrupted circadian rhythms have been described in Parkinson disease (PD) and other neurodegenerations [3-6], and some treatments specifically target circadian disruption [7]. But timing issues are not specific to neurodegenerative disease. A number of contemporary reviews examine motor symptoms in encephalitic syndromes with neural antibodies [8], the complex relationship between restlessness and DA [9], and dystonias characterized by diurnal variation and response to levodopa [10].
We feel that a single review would help to summarize entities that have been described in the past more or less individually. We offer a primer of practical, clinical basics regarding time- or state-dependent movement disorders. Although we attempt thoroughness, we do not claim to provide systematic reviews of the diseases we discuss. Our choice of citations involved different search strategies, depending on the disease category. For the antibody-mediated syndromes and for Willis-Ekbom (restless legs) syndrome, we consulted comprehensive reviews with the intention specifically to revisit original citations, and to highlight clinical and laboratory details not always provided in reviews. In other sections of this paper, we adopted more traditional, search-engine based reviews of the literature. We describe our search strategies in the “Supplementary Material” to this paper.
General Considerations from Chronobiology
Neuronal activity varies across light-dark cycles, particularly among the 10-20,000 specialized neurons of the paired suprachiasmatic nucleus (SCN) of hypothalamus [11-13]. SCN is considered the dominant circadian pacemaker in mammals. As the name indicates, it is located above the optic chiasm in anterior hypothalamus. Its neurons oscillate their resting membrane potentials and their firing rates, both of which are coupled to a 24-hour transcriptional-translational feedback loop, whose genes and associated proteins (e.g., Clock [Circadian Locomotor Output Cycles Kaput], Period1 [Per1], among many others; see Figure 1) have been studied in detail [12]. The view that SCN drives all circadian periodicities, however, has been challenged, based on observations that Per1 (inter alia) is expressed variously throughout the brain, that extra-SCN sites can manifest their own firing rhythms, and that multiple brain oscillators (or “clocks,” not referring to the “Clock” gene) contribute significantly to the control of physiological systems [11-14].
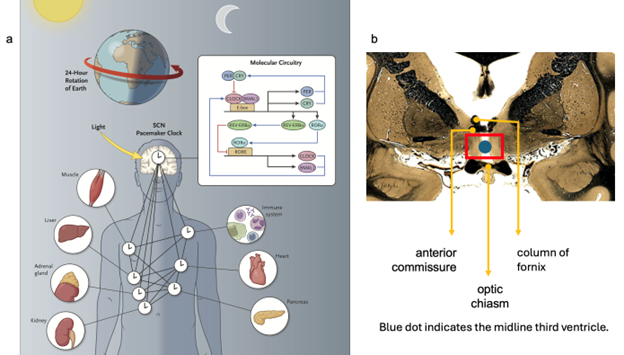
Figure 1: Some locations of biological clocks. 1a. The inset summarizes translational and transcriptional loops that operate in suprachiasmatic nucleus (SCN) of hypothalamus. PER: Period (clock repressor); CRY: Cryptochrome (clock repressor); CLOCK: Circadian Locomotor Output Cycles Kaput (core clock loop); BMAL1: Brain and Muscle “Arnt”-like Protein 1 (core clock loop); E-box: Enhancer Box; REV-ERB alpha: Member of Superfamily of Intracellular Transcription Factors; ROR: Retinoic Acid-Related Orphan Receptor (reinforces loop activity); RORE: ROR Response Element.
Note that not all “peripheral” clocks in the body or elsewhere in the brain (other than in hypothalamus) need to be synchronized with the SCN “master” clock. Figure from Allada and Bass (11), © 2021 Massachusetts Medical Society, with permission.
1b. Paired SCN(s) are located in the preoptic area of anterior hypothalamus (inside the red box), inferior to both the columns of the fornix and the anterior commissure and superior to the optic chiasm. A column of fornix, the midline anterior commissure, and the optic chiasm are indicated by orange arrows. The blue dot sits in the cavity of the third ventricle.
Coronal image of brain (fiber tracts stain darkly): courtesy of the National Museum of Health and Medicine, Michigan State University and the University of Wisconsin, available online at http://neurosciencelibrary.org. The anatomical collections are supported by the National Science Foundation and the National Institutes of Health, and are in the public domain.
Animal studies find that age (three months versus 24 months in normal mice) adds another consideration. In old mice, SCN neurons fire increasingly out of phase with each other when compared to young mice, although SCN neurons retain overall differences in oscillation between light and dark intervals of a day [15]. SCN is a dominant pacemaker, but not the only one in the brain, and the coherence of its neuronal firing varies over a lifetime. Furthermore, brain clocks like SCN differ from so-called “peripheral” clocks, which are present in cells throughout the body, and which govern diverse metabolic processes which themselves vary both over 24 hours and with age [11-14].
Even a cursory view of a rich chronobiological literature raises issues that frankly blur when we consider the time of day of neurological signs and symptoms. For example, “sundowning” colloquially refers to disruptive behavior during a transition in a 24-hour cycle [16]. But there is no consensus definition of the sundowning phenomenon [17]. Seeking cause for a change linked to time of day introduces a surfeit of possibilities that may coexist. Some major considerations, detailed in Table 1, speak to a widely accepted notion that there are many biological clocks and many ways by which those clocks, in both brain and body, can mis-time.
Returning to the idea of a dominant pacer: overall, light and dark difference tracks with motorically active and rest periodicity. If normal movement varies over a day, can we describe time dependency in movement disorders?
|
Factor |
Example |
|
Misalignment between SCN firing and the environment |
Night-shift work; travel across multiple time zones |
|
Misalignment between SCN as master clock and other brain or peripheral clocks |
See “Time-of-day studies in Parkinsonism,” below: disturbance of temporal cycles of activity and rest |
|
Exogenous or iatrogenic factors |
Use of psychotropic medications, e.g. for "sundowning" |
|
Circadian disruption promoting pathogenesis in distinct organs of the body |
Attenuated response to oxidative stress with consequent further disorganization of time perception [6,13] |
Time-of-Day Studies in Parkinsonism
Even before the development of motor signs, is there already a disturbance in cycles of activity and rest in Parkinsonism?
A behavioral disorder that manifests during rapid-eye-movement (REM) sleep is well known to be a herald of alpha-synucleinopathies, especially idiopathic Parkinson disease (PD), before the appearance of classical motor features of bradykinesia, rigidity, and tremor, or combinations of those three signs [18-20]. A phenoconversion rate from the polysomnographic diagnosis of an idiopathic REM-sleep behavior disorder (RBD) to an overt neurodegenerative syndrome (PD, dementia with Lewy bodies [DLB], multiple system atrophy [MSA]) has been quoted at roughly six percent per year, with overall ~75% conversion after 12 years [18].
Antelmi et al. [21] invoke criteria for diagnosis from the International Classification of Sleep Disorders, third edition: importantly, RBD requires polysomnographic demonstration of loss of muscle atonia during REM.
REM without atonia (RWA) is a kind of hyperkinesis, since muscle atonia is physiologic in REM. The absence of atonia in RWA is pathological by definition. Actigraphy, which enlists a watch-like action sensor worn at the wrist or ankle or both, is used in evaluations by sleep specialists, but video polysomnography remains the gold standard for evaluation of RWA. The value of Antelmi et al.’s review is its attention to a long list of idiopathic RBD mimics, some discussed later in this paper, but RBD in obstructive sleep apnea is especially common. As noted, idiopathic RBD carries high predictive value for an ensuing synucleinopathy, so exclusion of mimics has prognostic significance with implications for disease modifying interventions to mitigate progression in a parkinsonism.
Dream enactment in RWA, despite a connotation of dramatic action, can be subtle, and scoring of RWA relies on such nuances, which need not manifest during all REM epochs, and which can be very brief (e.g., three seconds in duration) [21]. Movements include tonic or phasic muscle activity of the chin (mentalis or submentalis muscle) and phasic activity of flexor digitorum superficialis (action of which involves flexion of the index finger through pinkie at the proximal interphalangeal joints) [22].
RBD antedates and commonly coexists with clinical Parkinsonism—in 60% of PD patients and in 80-100% of those with either DLB or MSA [23]. RBD may predict “malignant” forms of Parkinsonism characterized by more severe cognitive sequelae [24]. EMG phasic activity occurs only in REM sleep in RBD, consistent with absence of atonia in RWA [23]. We emphasize, however, that such EMG signatures offer only a snapshot of what can be far more striking movements in RBD.
The compelling observation of a parkinsonian prodrome specifically in sleep invites questions regarding pathophysiology in RWA and idiopathic RBD. Is DA deficiency or alpha-synuclein or both involved? A related question pertains to what is known in the time domain after parkinsonism manifests its hallmark clinical signs.
Does PD vary diurnally?
In a cross-sectional study of untreated (de novo) PD patients, van Wamelen et al. [25] evaluated 174 subjects from 7 a.m.-10 a.m., 181 from 10 a.m.-1 p.m., and 63 from 1 p.m. to 6 p.m. Using the Movement Disorders Society United Parkinson’s Disease Rating Scale, part III (MDS-UPDRS III), the authors found statistically significant worsening over that abbreviated day (7 a.m.-6 p.m.), especially in bradykinesia. The concern about de novo status pertains to the potential confound of any PD treatment, since both DA loss and dopaminergic treatment may affect endogenous timing mechanisms, in accord with an observation mentioned earlier, that DA neuronal firing constitutes a non-SCN clock [2]. Patients were almost exclusively in Hoehn and Yahr stages one and two (HY1, 2), viz., with either unilateral (HY1) or bilateral (HY2) signs without postural abnormality. Their MDS-UPDRS III scores varied from medians of 18 to 20 to 23 for the three consecutive time epochs, respectively. All the medians are relatively low, indicating mild-to-moderate motor impairment. An attempt to corroborate those data longitudinally in a smaller cohort (27 readouts in 12 subjects, who wore motion sensors until 9 p.m.), found worsening of bradykinesia from 9 a.m. to 9 p.m. When we compare van Wamelen et al.’s findings to an earlier, controlled investigation [26], variation also happens in age-matched controls, but the overall difference between the most and least active times of day was greater for controls than for patients, including those with advanced PD (HY3-5).
We conducted a targeted literature review to address what has otherwise been reported regarding daytime and nighttime variability in PD and, especially, PD manifestations at night (see “Supplementary Material” for our method). Treated PD patients have been studied using novel techniques, including smartphone and other actigraphy technology [27-29] and analysis of gait initiation disturbances [26]. Those studies report reduced amplitude of rest versus active time epochs in a day, and the findings have been corroborated in other studies of both de novo and advanced PD [reviewed in 4 and 30-32].
Nocturnal movements in Parkinsonism
Loss in the amplitude of difference between more and less active time epochs in a waking day does not speak to motor manifestations in sleep. Nor did our preceding discussion of RBD/RWA address what has been otherwise reported specifically at nighttime in PD.
Two thirds to nearly 100% of PD patients report insomnia [33,34]. Given the ubiquity of the problem, questions arise about what causes sleeplessness in PD, as multiple rating scales try to ascertain and to quantify [33,35]. An “insomnia syndrome” relates to difficulty falling asleep, awakening during the night, early awakening, and associated daytime fatigue related to lack of restful sleep. These syndromal criteria keep company with each other [36]. Specifically in terms of motor manifestations under the broad rubric of “tossing and turning,” Mirelman et al. [34] observe that overall “nocturnal lying time” (first >60 minutes supine to last 60 minutes supine) does not differ between patients and controls, but fragmentation of rest (greater upright time during the night) occurred in PD patients, with greater upright time associated with increasing PD severity.
Transition from a supine to upright position differs from restlessness in recumbency and movement disorders of sleep or resting wakefulness [36]. We discuss restless legs in a separate section dedicated to the Willis-Ekbom syndrome, below. Here we draw attention to restlessness and nocturnal movement specifically in PD. The restlessness, typically but not exclusively in the legs, is not the result of myalgia, cramping, venous insufficiency or other etiologies. Zhu et al. [37] and Matsubara et al. [38] interviewed a total of 325 PD patients to find that an urge to move with or without actual movement occurs in PD (when patients are supine but awake), sometimes with asymmetry (restlessness greater on the side first afflicted, since presentation in PD is characteristically asymmetrical). Although Matsubara et al. found that bilateral symptoms were more common, other studies [39-41] corroborate PD-like asymmetry. Neither Zhu et al. nor Matsubara et al. speak in detail about the nature of leg movements when they occurred (“walking” and “stretching” are mentioned).
Yet very specific motor phenomena have been described, including periodic leg movements of sleep (PLMS) and periodic leg movements of resting wakefulness (PLMW) [33,42]. The movements are involuntary and stereotyped: dorsal extension of a big toe together with partial flexion at the ankle, knee, and sometimes the hip; movements last from 0.5-10 secs with a run of at least four movements with variable inter-movement intervals, up to 90 secs [43]. We discuss PLMS and PLMW in greater detail in our discussion of Willis-Ekbom syndrome, below.
Studies in timing disruption in Parkinsonism
RBD is more likely in MSA or DLB than in PD—to which we can add that RBD is rare in Alzheimer disease (AD), uncommon in Huntington disease (HD), and also rare in most tauopathies except for a Guadeloupean tauopathy with features of a progressive supranuclear palsy (Gd-PSP) [23,44]. Once clinically diagnosed with a parkinsonism, sleep-associated motor manifestations—either RBD, PLMS/PLMW, or both—do not regress [23,43]. Can one localize a lesion or the lesions specifically responsible for the loss of atonia, the elaboration of RBD, or for the specific phenomenology of PLMS/PLMW in conditions associated with DA deficiency?
Nollet et al. [45] and French et al. [46] emphasize the redundancy of circuits involved in sleep-wake regulation, despite a “master” timekeeper (SCN of hypothalamus). Specific to DA, one anatomical approach is to think about where DA neurons (DANs) are in the brain. French et al. [46] discuss dopaminergic amacrine cells of retinas which input directly to SCN. Li et al. [47] and French et al. [46] discuss how SCN circadian genes and their protein products (e.g., Clock) both modulate and are modulated by DANs: 1. Clock gene expression directly affects both the rate-limiting enzyme for DA synthesis (tyrosine hydroxylase) and, therefore, DA metabolism at the level of the corpus striatum; 2. circadian genes effect DAN activity at the midbrain ventral tegmental area (VTA) probably by a post-translational mechanism; and 3. DA itself regulates Clock function in SCN. Nollet et al. [45] further describe how VTA in mice contributes to the generation of both non-REM and REM sleep, by way of differing projects of VTA DANs—for example, a VTA projection to basolateral amygdala in the generation of REM sleep, as was reported in 2022 [48].
Contemplating cell loss in PD—a neurodegeneration not restricted to DA nuclei—Mariotti et al. [49] argue that early involvement of locus ceruleus (LC), a catecholaminergic nucleus which perhaps degenerates before DAN nuclei, may specifically be responsible for RBD in PD. Rathor and Ch [50] observe that, although circadian disturbance occurs in genetic parkinsonisms in Drosophila, overexpression of alpha-synuclein has not been consistently associated with circadian disturbance. French et al. [46], however, convincingly argue that several brainstem and diencephalic nuclei—including brainstem locales associated with the ascending reticular activating system and orexin-producing cells of hypothalamus—are likely also involved in the evolution of sleep-related phenomena in Parkinsonism.
Various dopamine related mechanisms related to timing disturbances are summarized in Table 2.
|
Input from external world to SCN |
Amacrine cells of retina (altered transmission of environmental light) |
|
SCN and DA synthesis |
SCN gene expression regulates DA synthesis by action at rate-limiting enzymes of DA synthesis |
|
SCN and DA projections |
Post-translational effects: demonstrated, in particular, for ventral tegmental area of midbrain (a DA nucleus) and its projections to amygdala and corpus striatum |
|
DA effects on SCN |
DA itself regulates CLOCK (Circadian Locomotor Output Cycles Kaput) gene expression |
|
A role for neuroinflammation |
Microglial activation in Parkinsonism contributing to disturbed DA synthesis and abnormal SCN-DA interaction |
Neuroinflammation as causal?
Localization, as the above brief summary suggests, is not straightforward. The difficulty augments when we consider a striking thought offered by Li et al. [47]. The authors are not sure that circadian or other temporal disruption is necessarily the consequence of progression in PD, because circadian disruption may itself cause or exacerbate PD pathophysiology, as a result of neuroinflammation under non-SCN control in brain microglia [see also 50-52]. An inflammatory mechanism for sleep disturbance and motoric manifestations has precedent in autoimmunity associated with narcolepsy type 1 (autoimmune targeting of orexin containing neurons) [53].
We now turn our attention to well-characterized, antibody-associated syndromes in which disturbed consciousness or a sleep abnormality occurs or may eventually manifest in concert with a movement disorder. After our discussion of antibody-syndromes, we address Willis-Ekbom (restless legs) syndrome and selected dystonias with time-of-day manifestations which define them.
State (Sleep)-Related Movement Disorders in Antibody-Associated Disease: Major Examples
As we have seen, the Parkinsonisms exhibit disturbances in sleep. In a state of altered consciousness, as in an encephalitis, what non-Parkinsonian movement disorders are important to notice?
The movement disorder, if appreciated, clues one to diseases for which early diagnosis is vital. Early therapeutic intervention based on prompt diagnosis of antibody-associated encephalitic disease increases the likelihood of favorable outcomes. In this section, we address seven major antibody-associated encephalitides. Table 3 summarizes red flags for these disorders as well as laboratory correlations.
While excellent reviews have been published on the general topic of these entities [54,55], we focus on aspects at the intersection of abnormal sleep and movement, based primarily on first reports of a given syndrome.
|
Antibody mediated syndrome |
Red flag signs/symptoms |
Typical diagnostic findings |
|
Anti-IgLON5 |
RBD, gait-predominant parkinsonism and supranuclear gaze palsy, bulbar symptoms, dementia with craniofacial dyskinesia |
MRI: typically nonspecific [66]; some reports of diffusion restriction in high pontine tegmentum, dorsal midbrain, deep cerebellar white matter, superior cerebellar peduncles, and thalamus [67,68]; T2 hyperintensities can be seen in hypothalamus and tegmentum [69] |
|
EEG: nonspecific [66] |
||
|
CSF: cell count <10, total protein elevation (median 86mg/dL) [63,70] |
||
|
Anti-NMDAR |
Adults: Acute psychiatric changes, seizures, mouth and arm dyskinesias that persist in sleep. Antecedent HSV encephalitis [74]. Children: acute psychiatric changes, new insomnia, decreased level of consciousness, orofacial dyskinesias [86]. |
MRI: Findings not part of extant diagnostic criteria, but FLAIR changes have been seen in medial temporal lobes, basal ganglia, cerebellum, brainstem, hypothalamus, corpus callosum, and other nonspecific regions [76]. Atrophy can be seen in post-recovery phase [85]. |
|
EEG: focal or diffuse slowing, disorganized activity, “delta brush” [82] |
||
|
CSF: mild pleocytosis is common, range is high [77,83]; total protein can be elevated [76]; oligoclonal bands are common but may require multiple samples [83] |
||
|
Anti-CASPR2 |
Peripheral nerve hyperexcitability; fibrillary chorea; severe refractory insomnia with dysautonomia, seizures, encephalopathy, agrypnia excitata, RWA. Insidious development over months [88,89]. |
MRI: normal in 70% [94] |
|
EEG: epileptiform discharges, slowing [94], abnormal sleep including absent non-REM sleep architecture [93] |
||
|
CSF: normal in up to 65% of cases, can see mild pleocytosis or protein elevation [94] |
||
|
Other: EMG shows peripheral hyperexcitability [89,93,94] |
||
|
Anti-LGI1 |
Faciobrachial dystonic seizures (FBDS) [99-103], encephalopathy, amnesia, RBD with encephalitis, any combination of the above with concurrent hyponatremia |
MRI: medial temporal lobe FLAIR hyperintensities [92,93] |
|
EEG: epileptiform discharges, focal slowing in FBDS [99-103] |
||
|
CSF: typically normal, can have mild pleocytosis [98] |
||
|
Other: serum hyponatremia [99-103] |
||
|
Anti-Ma2 |
Hypothalamic/diencephalic dysfunction, limbic encephalitis, brainstem encephalitis, excessive daytime sleepiness with cataplexy, RBD with parkinsonism and vertical gaze palsy and/or apraxia of eyelid opening, RWA [80,107-110] |
MRI: FLAIR hyperintensities in medial temporal lobes and multiple subcortical regions have been described (thalamus to medulla) [108] |
|
EEG: abnormal sleep architecture [109] |
||
|
CSF: mild to moderate pleocytosis (5-113 cells/mliter), mild to moderate protein elevation (47-747mg/dL), reduced hypocretin levels [111] |
||
|
Other: commonly associated with cancer, including: lung, GI, breast, kidney, lymphoma, melanoma [105]. |
||
|
Anti-DPPX |
Classic triad: Weight loss with diarrhea, cognitive dysfunction, CNS hyperexcitability (exaggerated startle, myoclonus, seizures); PLMS, RBD, OSA, agrypnia excitata [112-115] |
MRI: normal or nonspecific [112] |
|
EEG: abnormal sleep architecture; report of simultaneous REM and non-REM architecture [114] |
||
|
CSF: mild to moderate pleocytosis (11-117 cells/mliter), mild protein elevation (up to 82 mg/dL) [112] |
||
|
Anti-GABABR |
New seizures, memory changes, behavioral abnormalities, excessive sleep or insomnia; one description of reduced REM sleep, fluctuating bulbar syndrome, and agrypnia excitata [116-123]
|
MRI: FLAIR hyperintensities in medial temporal lobes [120] |
|
EEG: normal or nonspecific |
||
|
CSF: mild to severe pleocytosis (0-950 cells/mliter) and mild to moderate protein elevation (35-109 mg/dL) |
||
|
Other: SCLC, other autoantibodies can be seen. Among those with SCLC – anti-SOX, anti-amphiphysin, anti-Hu/ANNA1, anti-Ri/ANNA2. Among those without SCLC – anti-GAD, anti-NMDAR, anti-IgLON5 [120-123]. |
||
|
We summarize relevant antibody-mediated syndromes with red flags and typical diagnostic findings. Note the concurrence of sleep and movement disorders and overlap between the syndromes. Different antibodies may lead to similar dysfunction of cortical and subcortical structures. CSF: Cerebrospinal Fluid; CNS: Central Nervous System; EEG: Electroencephalogram; EMG: Electromyogram; FLAIR: Fluid-attenuated Inversion Recovery; GI: Gastrointestinal; HSV: Herpes Simplex Virus; MRI: Magnetic Resonance Imaging; OSA: Obstructive Sleep Apnea; PLMS: Periodic Limb Movements Of Sleep; RBD: Rapid-Eye-Movement (REM) Sleep Behavior Disorder; SCLC: Small-Cell Lung Cancer |
||
Anti-IgLON5 (Immunoglobulin like, superfamily of five cell adhesion molecules, including limbic system-associated membrane protein, opiate-binding cell adhesion molecule, neuronal growth regulator 1, neurotrimin, and “IgLON”5) [56], antigen at plasma membrane
In Sabater et al.’s [57] 2014 report of eight patients, the authors reproduced detailed polysomnography for patient one, their index case, who was a 59-year-old man who presented with abnormal sleep behaviors, sleep apnea, stridor, hypersalivation, excessive sweating, and episodic but “intense” daytime sleepiness—all in the absence of dementia, parkinsonism, gaze palsy, gait disorder, ataxia, or daytime dyskinesia. Motor events occurred in REM sleep, “poorly structured” stage-two non-REM sleep, and in “undifferentiated” non-REM epochs (irregular theta, no vertex waves, K complexes, sleep spindles, or delta slowing). RBD was present based on recording of submentalis muscle and limb movements during REM sleep. In both REM and non-REM sleep, simple and complex vocalizations, jerks of multiple muscles at a time, kicking (although the leg movements did not meet criteria for PLMS), punching, and “finalistic” movements, which resembled identifiable activities such as eating or the manipulation of objects, were observed.
After obtaining cerebrospinal fluid (CSF) and serum, immunohistochemistry on sections of rat brain and cultured neurons identified an antibody to a cell-surface protein later determined to be IgLON5, based on study of transfected human epithelial kidney (HEK) cells. Given known human leucocyte antigen (HLA) associations with autoimmune disease generally, the authors pursued genotyping. Patient one and three others had HLA-DQB*05:01 and HLA-DRB1*10:01 alleles. Neuropathology in two patients (not patient one) found tau collections beyond what is seen in normal aging in hypothalamus, prehypothalamic area, and brainstem tegmentum (laterodorsal tegmentum, periaqueductal grey, pedunculopontine nucleus, magnocellular nucleus, and nucleus ambiguus).
Among the eight patients reported in 2014, four presented with a sleep disorder (one of whom had central hypoventilation) and four with non-parkinsonian gait instability (three of whom had central hypoventilation). Two of the latter four developed sleep-associated movements after the gait disturbance. All those with central hypoventilation died. Subsequent publications have elaborated a wide spectrum of presentations in anti-IgLON5 [58-70].
Gaig et al.’s [58] four fundamental clinical profiles help to organize the variability. At onset, anti-IgLON5 may manifest as 1.) a movement disorder in sleep, as in patient one, above; 2.) a bulbar syndrome that can resemble myasthenia gravis or motor neuron disease [64]; 3.) a gait disturbance (unsteadiness/imbalance, a feeling of lateropulsion or retropulsion), sometimes with vertical gaze palsies that might lead one to think about progressive supranuclear palsy (PSP, in which RBD is not at all common), but axial stiffness and rigidity presents only in ~25% [60]; and 4.) a cognitive disturbance with hyperkinesias (craniofacial movements commonly). For the last three, sleep disorders may evolve later in a patient’s course. Relationship to concomitant or antecedent infection (e.g., Epstein-Barr virus) is “plausible” [62], but has not been studied systematically. Cancer associations have been reported in up to 15%, but the discovery of tumors may be coincidental, based on diagnostic criteria for a paraneoplastic neurologic syndrome [65]. Standardized evaluation for malignancy was not part of one study (n = 20) in which breast adenocarcinoma in one patient was diagnosed prospectively [59].
Anti-IgLON5 treatment experience: Grüter et al. [63] distinguish between short-term immunotherapy (single courses of intravenous immunoglobulin (IVIg), IV steroids, or plasmapheresis [PLEX]) vs. long-term/long-acting immunotherapy (azathioprine, rituximab, pulsed cyclophosphamide, repetitive IVIg, relapse-independent use of steroids, or repetitive PLEX). In their retrospective review of 53 patients, early initiation of short-term therapy (less than six weeks after diagnosis) resulted in physician-judged improvement. Long-term therapy resulted in response up to 75%. Serum or CSF anti-IgLON5 titers rose with increasing time after diagnosis, despite treatments. Mortality, often due to respiratory complications, reached 19% after three years.
Pathophysiologic questions raised by anti-IgLON5: As Giannoccaro et al. have discussed [71], anti-IgLON5 specifically raises mechanistic questions at the interface of neuroimmunology and neurodegeneration. We summarize such questions (and answers) in Table 4. Regarding other antibody syndromes, answers are less clear, due to variability in reports or lack of information to date.
|
Question |
Answer, for Anti-IgLON5 |
|
Red flag for syndrome? |
RBD, among others |
|
Nature of antibody (Ab)? |
Cell surface antigenic target; IgG4 >> IgG1 |
|
Does Ab appear in primary neurodegenerations? |
No, compared to anti-NMDAR (IgM, IgA, or IgG) present in ~15% of dementias |
|
Are there biomarkers or predictors of possible therapeutic response? |
Yes, lower serum neurofilament light predicts response to long-term treatment |
|
Receptor cross-linking at cell surface, with internalization? |
Yes |
|
Binding disruption at surface? |
No, compared to interaction with “A Disintegrin and Metalloproteinase domain 22” (ADAM22) in anti-LGI1, with subsequent cellular internalization of ADAM22-anti-LGI1 |
|
Functional blockage of antigenic target? |
No, compared to anti-GABAB. Internalization of surface anti-IgLON5 with ensuing neurodegeneration |
|
Irreversible internalization of antigen? |
Yes |
|
Evidence for complement activation or other inflammatory factors? |
Yes, in animal models: increase in TGF-beta (tumor growth factor-beta), chemokines |
|
Evidence for B- or T-cell activation? |
Yes, increases in B-cell numbers in CSF |
|
Evidence for other inflammation (microglial or astrocytic activation; cytokine mediation)? |
Some evidence (lymphocyte infiltration, microglial activation) |
|
HLA associations? |
Yes, as discussed in text |
|
Neuropathological correlations? |
Hyperphosphorylated tau in neurons (hypothalamus, brainstem tegmentum) with neuronal loss—but not all studies concur |
Anti-NMDAR (N-methyl-D-aspartate—NR1 and NR2 subunits—receptor), antigen at plasma membrane
Since its discovery, anti-NMDAR has become recognized as the most common non-viral encephalitis [72-86]. Nine young women reported by Vitaliani et al. all had ovarian teratomas 74). Armangué et al. [75] describe that antecedent herpes simplex virus (type 1) encephalitis (HSVE) may trigger anti-NMDAR. Progression in anti-NMDAR is common. In adults, psychiatric symptoms happen early. A decreased level of consciousness and dysautonomia happen later [76-78]. Female predominance has been confirmed in multiple studies [72,78]. Children are more likely to present with irritability, insomnia, abnormal movements, and seizures, while adults tend to present with psychosis, and are more likely to develop hypoventilation [72,78,79].
A common anti-NMDAR sleep disturbance is insomnia with daytime hypersomnolence [76,80]. What has been described as status dissociatus/agrypnia excitata (to be discussed in greater detail with respect to anti-CASPR2, below) may present with complex movements (antigravity in the arms especially, as well as perioral dyskinesia) which can be evoked with sensory stimulation, and which occur in “a state of paradoxical and alternating responsiveness [81].” Dream enactment and disorders of arousal (“confusional arousals and somniloquy”) occur, as does RWA [76,80]. Sleep disturbances can persist despite treatment. Interestingly, the hypoventilation that is a hallmark of more advanced encephalitis is not always associated with a decreased level of consciousness or can manifest only during sleep [80].
Enhanced recognition of encephalitides in general has led to the development of diagnostic criteria specific for anti-NMDAR [82]: onset over less than three months of at least four “major” symptoms (e.g., abnormal behavior/cognition, a movement disorder especially in the context of a decreased level of consciousness, central hypoventilation, others); at least one of two laboratory features (abnormal EEG or CSF pleocytosis); and the exclusion of other diagnostic possibilities. In anti-NMDAR, EEG changes (focal or diffusely slow or disorganized activity, “delta brush,” a variant of delta rhythm in which the top of a slow and wide EEG waveform has the appearance of bristles of a brush) are common, even in the absence of seizure or frank epileptiform activity. CSF pleocytosis or oligoclonal bands (OGBs) are part of the criteria and are common findings.
Anti-NMDAR treatment considerations: Search for a teratoma is indicated in suspected anti-NMDAR. Anti-NMDAR is a treatable encephalitis. Roughly 80% improve with immunotherapy (first- or second-line agents) and/or oncologic treatment, although recovery may be slow [72].
In an archival autopsy cohort, two untreated anti-NMDAR cases (one with comorbid small-cell lung carcinoma [SCLC]) and two additional cases (one with coexisting demyelination and another with a post-transplant lymphoproliferative disorder) were studied [87]. Of note, in the absence of immune treatment, the clinical triad of a movement disorder, abnormal behavior, and memory dysfunction correlated with plasma cell inflammation of basal ganglia, amygdala, hippocampus. The findings corroborate an antibody-mediated mechanism for the development of both dyskinesia and psychiatric/cognitive disturbance in anti-NMDAR.
Anti-CASPR2 (Contactin-Associated Protein-like 2), antigen at plasma membrane
CASPR2 is a transmembrane protein, one of two major voltage-gated potassium channel (VGKC)-associated antigenic targets. We discuss the second antigen, leucine-rich glioma inactivated 1 (LGI1), below. Discovery of anti-CASPR2, both as syndrome and autoantibody, advanced understanding of pathophysiology in neuromyotonia and Morvan’s syndrome (the latter almost always in men, manifesting as the triad of encephalopathy, dysautonomia, and peripheral nerve hyperexcitability) [88-96]. We include a discussion of early historical reports, like Morvan’s, in “Supplementary Material.”
Compared to anti-LGI1, anti-CASPR2 is more likely to have a coexisting tumor (thymoma most commonly) as well as a spectrum of ongoing motor discharges, including neuromyotonia, myokymia, cramps, fasciculations, myoclonus in the legs on standing, or a low amplitude, very high frequency, and arrhythmic movement disorder described by Morvan as “fibrillary chorea.”
Acquired neuromyotonia and Morvan’s syndrome were recognized entities prior to the discovery of anti-CASPR2 antibodies. The clinical spectrum of anti-CASPR2 includes both, along with other phenotypes. Van Sonderen et al. [89] retrospectively analyzed a Spanish and Dutch cohort (n = 38) with serum or CSF positivity for anti-CASPR2. They identified seven “core” symptoms: cerebral symptoms, cerebellar symptoms, peripheral nerve hyperexcitability (including myotonia), dysautonomia, neuropathic pain, weight loss, and insomnia.
The last symptom is notable. A syndrome termed agrypnia excitata [90-92] (a “chase” for sleep, with motor overactivity—characterized by severe insomnia and “oneiric stupor” [hallucinations, motor agitation]), various dysautonomias (fever, perspiration, tachycardia, tachypnea, hypertension), and other motor activity including RWA—are all hallmark features of anti-CASPR2. Agrypnia excitata, however, occurs in other antibody syndromes such as anti-NMDAR (see above).
Considered a type of status dissociatus (a breakdown in EEG distinctions between wakefulness, sleep stages, and transitions to sleep or wakefulness), agrypnia excitata polysomnographically is a “persistent state of subwakefulness . . . regardless of circadian rhythms” [21]. Liguori et al. [93] reported a case of a 79-year-old man with Morvan syndrome and VGKC antibodies whose polysomnography showed absent non-REM sleep architecture (sleep spindles, K-complexes, and delta waves were undetectable); atypically short REM epochs with RWA; and behavioral oscillation between calmness and agitation with dream enactment. Sleep architecture returned to near-normal after PLEX, and brain pathology showed only a microinfarct in the hippocampus without thalamic or brainstem changes.
Anti-CASPR2 treatment considerations: Anti-CASPR2 responds to treatment, including resection of thymoma if the tumor is detected [95]. Van Sonderen et al. [89] found that among patients without tumor, a majority showed full or partial response to immunotherapy. Shivaram et al. [96] reported eight patients who were treated with IV steroids and PLEX acutely, followed by pulsed IV steroids (a “long-acting” therapy, to use Grüter et al.’s [63] terminology). One patient died with refractory myasthenia with thymoma, but seven recovered “completely” after a mean follow-up period of ~20 months.
Anti-LGI1, antigen at plasma membrane
What are termed anti-VGKCs [97] in fact target LGI1 and CASPR2 specifically, as Irani et al. elucidated [88]. When we compare anti-LGI1 patients to those with anti-CASPR2, differences in their respective syndromes emerge.
Anti-LGI1, compared to anti-CASPR2, more commonly presents with amnesia, confusion, seizures, medial temporal lobe MRI findings, and, notably, hyponatremia (<135mM). As discussed above, anti-CASPR2 is associated with status dissociatus and agrypnia excitata, whereas anti-LGI1, albeit with various sleep disturbances [92,93], is more associated with RBD and PLMS.
Evidence for inflammation in anti-LGI1 CSF is often underwhelming [98].
Faciobrachial dystonic seizures (FBDS): We summarize five citations related to a distinctive movement disorder/seizure in anti-LGI1 [99-103].
FBDS are brief (lasting several seconds) and dystonic in appearance; they occur often in a state of reduced consciousness; a given paroxysm involves an arm and ipsilateral face in a stereotyped sequence. One hemibody is affected at a time, but the body side can alternate. Loss of awareness happens in the majority (66%); at onset, patients may experience “thermal” sensations, limb automatisms, and speech arrest. Epileptiform or slowed EEG waveforms appear in 24%. FBDS may manifest before the development of a full amnesic or confusional state or other evidence for encephalopathy or encephalitis. FBDS do not respond to antiseizure medications, but they do to immune therapies in 50%, roughly within 30 days after initiation of treatment (most commonly IV steroids, although antiseizure medications were given concomitantly in a prospective trial [103]).
After 90 days of active FBDS, 56% develop cognitive impairment associated with both evidence of hippocampal structural and functional change as well as higher proportions of complement-fixing IgG1 and IgG4 antibodies. As we mention passingly in Table 4, a combination of complement deposition and cellular internalization of ADAM22-IgG1-LGI1 (or ADAM22-IgG4-LGI1) complexes has been implicated in the pathogenesis of cognitive impairment, especially after delayed immune treatment for FBDS [99].
Anti-LGI1 treatment considerations: Anti-LGI1 responds to immunotherapy; there is a paucity of tumor associations. In Irani et al.’s series [88], anti-LGI1 had a significantly more robust response to immunotherapy (steroids, particularly) than anti-CASPR2. Early immune treatment may mitigate long-term cognitive sequelae.
Anti-Ma2, nuclear or cytoplasmic antigen
The Ma family of proteins is expressed in the brain and testes, both organs with a blood-tissue barrier. Cells expressing Ma have been studied in relationship to cancers, including melanoma and testicular germ cell tumors. Paraneoplastic Ma antigens (PNMAs; PNMA1 and PNMA2 in particular) are targets for two antibodies, anti-Ta (which recognizes PNMA2; anti-Ma2 is synonymous with anti-Ta [104]) and anti-Ma (which recognizes both PNMA1 and PNMA2). As Hoffman et al. [105] have described, young men with encephalitis and testicular germ cell tumors express anti-Ta/anti-Ma2, whereas patients of either sex—with a range of tumors—may express anti-Ma. We concentrate on the clinical picture associated with anti-Ma2.
Ten patients studied by Voltz et al. [106] had both anti-Ma2 and testicular cancer, with varied presentations. Five had seizures and six had memory loss (four had both seizures and memory loss). Hallucinations, abnormal eye movements (diplopia, oscillopsia), dysarthria, hypersomnolence, and ataxia were other features. Patient one presented with a syndrome of resting tremor, depression, and dystonia in addition to seizures and a memory disturbance. Patient ten manifested a mixed hypothalamic and parkinsonian syndrome with loss of libido, mutism, hypothyroidism, diabetes insipidus, and hypersomnolence.
In the early 2000’s [107,108], 38 cases of anti-Ma2 were characterized in detail. The majority presented with a combination of limbic, brainstem, and/or diencephalic encephalitis. Notable aspects included eye movement abnormalities (vertical gaze paresis was most common; non-paretic eye closure in a parkinsonian presentation resembled an apraxia of eyelid opening) and diencephalic dysfunction (13/38; features included excessive daytime sleepiness associated with low CSF hypocretin, cataplexy, hypnagogic hallucinations, unexplained weight gain, hyperthermia, and various endocrine disturbances related to hypothalamic-pituitary dysfunction).
In reviews of sleep disorders associated with neurological autoimmunity [80,109-111], anti-Ma2 has also been associated with excessive daytime sleepiness with cataplexy and low hypocretin/orexin, RWA, reduced sleep efficiency, increased sleep fragmentation, reduced sleep latency, absent sleep spindles, and absent slow wave sleep—the latter two aspects as seen in agrypnia excitata.
MRI (mainly T2/FLAIR) and CSF abnormalities are common in anti-Ma2 [108]. Multiple subcortical areas, from the thalamus superiorly to the medulla inferiorly, demonstrated lesions; medial temporal lobes structures were the most commonly involved cortical areas. The authors also reported on biopsy and autopsy material (they found T-cell inflammation commonly and multifocally), but we draw attention to a later study by Dauvilliers et al. [110] of a 63-year-old man with diencephalic encephalitis and anti-Ma antibodies (strong anti-PMNA2, weak anti-PMNA1 reactivity) with “almost exclusive inflammation and tissue injury in the hypothalamus.”
Anti-Ma2 treatment considerations: Over half of anti-Ma2 cases are associated with cancer, testicular tumors most commonly. In Dalmau et al.’s series of 38 patients [108], most received both oncologic (including orchiectomy) and immune therapy: 18 patients improved neurologically; 15 deteriorated. Younger men with testicular tumors are more likely to improve than those with non-testicular tumors [108,109].
In anti-Ma (with antibodies directed at both PNMA1 and PNMA2) Hoffmann et al. [105] reported that nine of 14 cases (eight female, six male) had histologically confirmed tumors—in lung, gastrointestinal tract, breast, and kidney, in addition to non-Hodgkin lymphoma and melanoma. In 13 of the 14, neurological disease progressed despite immunotherapy, although tumor disease stabilized with treatment in seven cases.
Anti-DPPX (Dipeptidyl-Peptidase-like-Protein 6), antigen at plasma membrane
In 2013, Boronat et al. [112] reported four patients with subacute encephalopathy with agitation, hallucinations, myoclonic jerks, and seizures. Three experienced a prodrome of severe, unexplained diarrhea and weight loss. Serum and CSF harbored antibodies directed against a 100 kD regulatory subunit (DPP6 or DDPX) of Kv4.2 (rapidly inactivating) VGKCs. DPPX is present on the cell surfaces of cultured hippocampal neurons as well as in the cytoplasm and on the membranes of myenteric plexus neurons. As with LGI1 and CASPR2 antibodies, DPPX antibodies are highly specific for the DPPX protein and not VGKC or adjacent proteins.
Oliver Tobin et al. [113], reporting on 20 anti-DPPX patients, noted a memory disorder in 16 of the 20 patients, various symptoms attributed to brainstem dysfunction (diplopia/blurred vision/oscillopsia in eight; dysphagia in six); myoclonus (in eight), diffuse rigidity (in six), exaggerated startle/hyperekplexia (in six), and hyperreflexia (in six). Prodromal weight loss was common (in 12), but not solely related to diarrhea; five patients presented with anorexia. Thermoregulation, cardiac conduction, and bladder disturbances occurred in patient #’s one, three, and seven, respectively. Seizures were uncommon (two patients). Tumor screening discovered B-cell neoplasms in two of the 20.
Sleep manifestations were frequent. Nine reported sleep “disruption” (six with insomnia); PLMS occurred in five. One patient studied polysomnographically evinced simultaneous REM (sawtooth EEG waves) and non-REM sleep (sleep spindles, K complexes)—a mixed picture consistent with status dissociatus. A subsequent 2023 review [114], with focus on sleep disorders in anti-DPPX in the Mayo Clinic experience, draws attention to RWA, RBD, obstructive sleep apnea, as well as the case of status dissociatus just mentioned; complete polysomnography was available for only three in their small sample (n = 6).
Hara et al. [115], reporting on 39 patients from 2013-2017 (nine newly reported in their paper), found that 67% overall presented with a triad of weight loss/diarrhea, cognitive/memory dysfunction, and CNS “hyperexcitability” (exaggerated startle, myoclonus, tremor, or seizures).
Anti-DPPX treatment considerations: Anti-DPPX responds to immunotherapy, as is true for other surface-directed antibody syndromes. In Hara et al.’s [115] review, overall improvement was seen in 60% (for their new nine cases, modified Rankin scores were no greater than two—suggesting modest impairment—in seven cases, after 12-36 months of follow up).
Anti-GABABR (Gamma-aminobutyric Acid “B” Receptor, B1 subunit), antigen at plasma membrane
A previously healthy 55-year-old woman [116] developed diplopia and fluctuating bilateral ptosis, then, two years later, dysphagia and tongue weakness. Three years after presentation, polysomnography revealed a reduction in REM sleep. Seven years after the presentation, she experienced episodes lasting a few hours characterized by early, heavy sweating, nocturnal respiratory crises, then respiratory failure. At one point during these bouts, she experienced 24-48 hours of fasciculations, cramps, and hyperreflexia. EMG found no evidence for ongoing motor activity. Whole-body MRI found no tumor, and her brain MRI was normal, as was her CSF. Various polysomnographic studies found evidence of agrypnia excitata with finalistic movements of the type described in our discussion of anti-IgLON5. During her protracted illness, improvement in sleep architecture and other aspects of her episodes was credited to various immune-modulating trials.
In anti-GABABR, sleep + movement disorders have been poorly characterized if reported (except the description provided above by one group [116,117]). Zhu et al. [118] conducted a retrospective analysis of 14 Chinese patients with anti-GABABR: abnormal sleep patterns occurred for six; “[four] patients slept more, whereas two were restless and did not sleep at night.” No polysomnography was reported. Similarly, Guan et al. [119] retrospectively studied 18 Chinese patients: three had a “sleep disorder,” with no further description of specifics.
Lancaster et al. [120] published a series of 15 patients, all of whom had seizures as part of their encephalitic syndrome. Cancer was common: 7/15 had an identified tumor, most commonly SCLC. One patient presented with “abnormal sleeping habits,” but details were not reported.
Höftberger et al. [121] reported 20 patients with anti-GABABR: the general presentation of seizures, memory loss, confusion, and the presence of SCLC echoes Lancaster et al. But they also observed ataxia, opsoclonus/myoclonus, and the presence of additional antibodies: among those with SCLC, anti-SOX (Sex-determining region Y-related HMG-Box [high mobility group Box]), anti-amphiphysin, and anti-Ri (or anti-ANNA2, antineuronal nuclear antibody type 2)—SOX, amphiphysin, and Ri/ANNA2 are all nuclear or cytoplasmic antigens. Among those without SCLC, anti-glutamic acid decarboxylase (GAD) and anti-NMDAR (NR1 subunit) were observed in addition to anti-GABABR. No sleep abnormalities were reported.
The association with other antibodies is notable. In Lancaster et al.’s series [120], 3/15 had both anti-GABABR and anti-GAD. Bonorat et al. [122] found, in association with SCLC and anti-GABABR (n = 8), that one patient co-expressed anti-Hu (anti-ANNA1) and two anti-GAD. In a single case of “clearly . . . ‘classical’” anti-IgLON5, with a gait disorder and severe parasomnias, anti-GABABR was present in addition to anti-IgLON5 [123].
Anti-GABABR treatment considerations: Anti-GABABR responds to immunotherapy and concurrent treatment of tumor (viz., SCLC). In a systematic review, McKay et al. [124] reported an 86% improvement rate with immunotherapy and relevant oncologic therapy in anti-GABABR.
Mimicry in Antibody-Associated Disease
Identification of an antibody may result in phenotype “creep”—the thought or presumption that a syndrome’s spectrum should broaden because of an unexpected antibody titer [125]. In the case of mimicry, an analogous problem arises: if, for example, one clinically suspects anti-NMDAR or anti-LGI1 (e.g., with oral dyskinesias), then what significance accrues to an unexpected antibody titer? We discuss three examples of antibody syndromes that mimic others.
Anti-Neurexin-3-alpha, antigen at plasma membrane
In five cases reported by Gresa-Arribas et al. [126], four were women; the clinical picture was consistent. A young person (<50 years) experiences a rapid decline in level of consciousness with seizures, subtle orofacial dyskinesias, and central hypoventilation. Two of the five died and the other three only partially recovered despite treatment. All three displayed ongoing cognitive deficits, though follow-up was variable (2-36 months). All patients received high-dose corticosteroids with one additionally receiving IVIg and another cyclophosphamide. Sustained remission was observed in a single case report of a 59-year-old woman with new-onset seizures, hallucinations, and aphasia who was found to have anti-neurexin-3-alpha antibodies in CSF. She was initially treated with high-dose corticosteroids and plasmapheresis, followed by a 1-year course of rituximab [127]. This patient had no residual cognitive deficits at 3 and 6 months. The presentation calls to mind either anti-NMDAR and anti-IgLON5 (orofacial dyskinesia and central hypoventilation occur in both), but Gresa-Arribas et al. found expression of anti-Neurexin-3-alpha in all cases. Neurexins are polymorphic neuronal surface proteins that are involved in synapse formation and maturation.
Anti-ZSCAN1 (Zinc finger- and Scan domain-containing protein 1), nuclear or cytoplasmic antigen
Based on a proteome-wide screen, a rare pediatric syndrome of rapidly developing obesity, hypothalamic dysfunction, hypoventilation, and autonomic dysfunction (ROHHAD) has been associated with expression of anti-ZSCAN (in 7/9 ROHHAD cases) and with neuroblastic tumors (in 8/9) [128]. ZSCAN is expressed particularly in hypothalamus—reminiscent of anti-Ma2, in which hypothalamic-pituitary dysfunction is a hallmark.
“Basal ganglia antibodies” and Anti-D2R (DA-2 Receptor), receptor at plasma membrane
In 2001, Dale et al. reported a dyskinetic form of basal ganglionic encephalitis resembling von Economo’s encephalitis lethargica in 10 children with prior streptococcal infection [129]. In 2009, they gathered CSF and serum in 20 cases, not all of whom had known streptococcal infection, to discover that the dyskinetic phenotype of encephalitis (with sleep disorder and psychosis) tested positively for anti-NMDAR in 10 [130]. In 2012, 17 children, again with basal ganglionic encephalitis, were compared to groups with Sydenham chorea, Tourette syndrome, and pediatric autoimmune neuropsychiatric disorder associated with streptococcal infection (PANDAS) [131]. Twelve of the 17 tested positive for anti-D2R, but also 10/30 with Sydenham and 4/44 with Tourette, but 0/22 with PANDAS.
Even in the early 2001 report, the research group suspected autoimmunity directed against “basal ganglia,” and an antibody specific to the DA-2 receptor corroborates that view, but the papers as a group speak to the possibility that syndromes (anti-NMDAR and anti-D2R) can mimic each other. Indeed, in a study of 48 pediatric cases of first-break psychosis, eight elaborated antibodies—three with anti-D2R, five with anti-NMDAR, and one with both anti-D2R and anti-NMDAR [132]. Likewise, in a pediatric sample studying chorea and encephalopathy following HSVE, either anti-NMDAR, or anti-D2R, or both anti-NMDAR and anti-D2R has been reported [133].
Restlessness in a State of Repose: Willis-Ekbom Syndrome
In the 17th century, Thomas Willis described an affliction of motor restlessness when persons “betake themselves to sleep”; Ekbom reported a syndrome of discontinuous paresthesiae associated with a need to move. For a discussion of historical descriptions of Willis-Ekbom syndrome, please see “Supplementary Material.”
Based on a 2014 consensus statement [134], criteria for Restless Legs Syndrome (RLS) include features not significantly different from Willis’s and Ekbom’s descriptions of akathisia as well as features “not essential” for diagnosis, despite their very close association with RLS, including: 1. PLMS; 2. PLMW; 3. DA treatment response, although we add that treatment options are myriad [135]; 4. a family history of RLS among first degree relatives; and 5. a lack of “profound” daytime sleepiness. Högl and Stefani [43] elaborate on those aspects, then broaden our view of RLS as a movement-disorder comorbidity.
RLS and PLMS, the latter diagnosis based on EMG criteria mentioned earlier, commonly occur together, although either PLMS or PLMW can occur without RLS. PLMS in isolation could be a biomarker of genetic susceptibility to RLS; linkages based on genome-wide association studies are summarized in a recent and exhaustive review of RLS by Manconi et al. [136]. Furthermore, both RLS and PLMS frequently manifest in other movement disorders—here we summarize Högl and Stefani’s long list: various Parkinsonisms (PD, MSA, DLB), tauopathies (PSP and corticobasal syndrome), essential tremor, Tourette syndrome (associated with maternal, not paternal RLS), various dystonias (cervical dystonia, blepharospasm, some generalized dystonias), Friedreich and other ataxias (especially spinocerebellar ataxia types 2 and 3 [SCA2 and SCA3]), propriospinal myoclonus of sleep onset, and excessive fragmentary myoclonus of sleep (“ubiquitously” comorbid with RLS).
A DA paradox?
Ferré et al. [9] describe a seeming DA paradox in RLS. Acute DA receptor antagonism commonly results in akathisia during wakefulness (an untoward neuroleptic side effect, which can also develop in tardive fashion). On the other hand, RLS, an akathisia, is a nocturnal phenomenon or at least one in recumbency, as is PLMS (for clarity, periodic leg movements of sleep), and both conditions respond to dopaminergic treatment. But prolonged use of DA agonists or high doses of those medications can result in “augmentation,” a worsening of RLS during treatment. Studies over time have built an argument for increased DA synthesis in untreated RLS [137].
The DA “paradox” has treatment implications. Higher doses of DA agonists can augment RLS severity but attempts to decrease or to discontinue the agonist are not tolerated, precisely because the restlessness persists or worsens. In such situations, transitions to other classes of drug (e.g., opiates) may be necessary, but it is often the case that combination treatment is necessary (e.g., a lower dose of agonist plus an opiate) [138].
DA tone in context; genetics and iron in RLS
Traditionally, DA has been implicated in the modulation of cortically driven movement, and DA’s role in motivation and learning, both in terms of a midbrain DA neuron’s tonic or phasic firing (phasic in very short time frames, over seconds) has been investigated in detail [139]. While there are suggestions that hypothalamic and striatal DA vary over the course of a day, the mean rate of midbrain DA neuronal firing may not [140,141]—the last point has been debated [142]. Nevertheless, DA “activity” in different parts of the brain, particularly the corpus striatum, to which midbrain DA neurons project, may vary over 24 hours as a function of multiple factors—e.g., the activity of DA transporters [141], modulation of DA action by glutamate and acetylcholine, or control mediated by DA and adenosine receptors especially in ventral corpus striatum [9].
Allen [143] has authoritatively discussed the related roles of genetics and of brain iron stores in RLS. Among several risk alleles (see also 136), BTBD9 on chromosome 6p is associated with RLS, PLMS independently of RLS, and decreased peripheral iron, as occurs in iron-deficiency anemia. Treatment of peripheral iron deficiency with decreased serum ferritin is well known to be effective in RLS, and RLS prevalence is roughly nine times greater in iron-deficiency anemia than in the general population.
A threshold of critical post-synaptic DA signaling exists, Allen has maintained; below it, RLS symptoms manifest. In patients who respond well to dopaminergic treatment, DA “activity” varies during the day without dipping below that threshold, with less at night and more during waking hours. In augmentation, ongoing treatment with dopaminergic agents results in larger swings in DA activity (more DA “activity” during the day with a nadir at nighttime), with more time spent below threshold and greater RLS symptomatology as a consequence. Low dosing and longer drug half-lives mitigate the problem.
Spinal excitability
Before 2020, reports had surfaced that a small diencephalic cluster of DA neurons—called A11 located in hypothalamus, dorsal to mesencephalic “A” groups [144]—projects densely to all Rexed laminae of spinal cord, especially to the sensory dorsal horn and intermediolateral grey matter [145-148].
Evolutionarily conserved, A11 contributes to modulation of spinal motor networks, perhaps by inhibition of sensory afferents [145]. A11 projections descend through periaqueductal grey to terminate more densely in lumbar than in higher levels of spinal cord [146], and A11 lesioned rats are hyperactive with long-lasting reduction in sensory thresholds [147].
Spinal cord disruption, in a decerebrate animal with transection at the level of the midbrain colliculi or in humans with cord injury, results in periodic leg movements that resemble both the triple flexion response (flexion at the ankle, knee, and hip) or various combinations of movements in PLMS or PLMW (for clarity: PLMW = periodic leg movements or resting wakefulness; both PLMS and PLMW exhibit extension of the big toe, partial flexion at the ankle, knee, and hip) in response to mechanosensitive and/or nociceptive afferents; disinhibition of flexor reflexes also occurs in the context of intact supraspinal influence, specifically in rest and sleep [148].
Ferré et al. [149] note that triple flexion, PLMS, and PLMW may all involve the same spinal “generator.” Hypoactive adenosinergic inhibition of that generator, if indeed causal, introduces novel therapeutic options, including use of adenosine agents such as dipyridamole in the treatment of the movement disorder.
Whether DA in spinal cord influences co-contraction of flexors and extensors is best addressed in a discussion of dystonias with diurnal variation.
Time-of-Day Variation in Dystonia
Segawa et al.’s original 1976 report [150] of “marked diurnal fluctuation in dystonia” remains a compelling description of a phenotype: typical onset in childhood, no birth or developmental abnormality, insidious onset of dystonia or other dyskinesia, progression typically over five years, a pattern of bodily involvement (if a leg is first involved, then the ipsilateral arm becomes dystonic, then the contralateral leg, then contralateral arm), marked diurnal variation (worse in the morning), and compelling response to low doses of levodopa. Recent reviews discuss many variant presentations, “look alikes,” and “dystonia plus” syndromes [151,152]. We undertook a review to summarize the many dystonias associated with single gene mutations that characteristically vary with the time of day. We describe our literature search method in “Supplementary Material” to this paper. Table 5 summarizes our findings.
Relevance of tetrahydrobiopterin (BH4) and DA pathways (Figure 2)
We schematize the synthesis, salvage, and recycling of BH4, an essential cofactor for the rate-limiting enzyme TH; in Figure 2. We also address the relevance of other gene associations, as cited in Table 5.

Figure 2: Relevance of BH4 and DA pathways; other relevant gene associations.
|
First author, reference number |
Key Findings |
|
Stephen [10,153], DePablo-Fernandez [154] |
DYT/PARK-GCH1: DRD, autosomal dominant (AD), rarely autosomal recessive (AR), dopa responsive, with diurnal variation (worse in evenings), sleep benefit, associated with spasticity and familial cerebral palsy; normal dopamine transporter (DAT) studies; Atypical features include parkinsonism in adult presentations, myoclonus, and various psychiatric comorbidities, including obsessive-compulsive disorder Marked diurnal variation characterizes DRD. An early sign is a distal dystonic posture in the leg, a pes equinovarus in a young person, more commonly among females. DYT/PARK-TH: DRD, AR, dopa responsive, with diurnal variation, associated with myoclonus, spasticity, oculogyric crises; deep brain stimulation (DBS) of benefit. DYT/PARK-SPR: DRD, AR, rarely AD, dopa responsive, developmental delay, high CSF biopterin/dihydrobiopterin. Additional rare AR form involves gene encoding PTP synthase. MxMD-ADCY5: “mixed” refers to its clinical pleiotropy: dystonia, chorea, myoclonus, tremor, triggered by transitions in sleep. DYT-DDC: AR, diurnal variation, sleep disorder, sleep benefit, chorea, developmental delay, truncal hypotonia, oculogyric crises, ptosis, autonomic symptoms |
|
Keller Sarimento [155], Wong [156] |
New genes responsible for DRD DnaJC12: bi-allelic loss-of-function mutations cause developmental delay, hyperphenylalaninemia, with favorable response to BH4 and levodopa SLC18A2: infantile onset DYT/PARK; dopamine agonists effective, but not levodopa Wong describes other defects in BH4 metabolism (DHPR, PCD); also describes posterior leukomalacia in DYT/PARK-GCH1 |
|
Carecchio [157] |
PDE10A related disorders: both AD and AR; carriers of dominant mutations with homogenous phenotype: early onset (<15 years old), chorea, normal cognition; diurnal fluctuations in childhood with progressive spread of chorea during life. |
|
Pérez-Dueñas [158] |
Periodic dystonic attacks, often associated with orolingual dyskinesia, with diurnal variation (with sleep benefit) in SLC6A3. ATP1A3: paroxysmal paralysis, dystonia, ataxia, and dysphagia; fatigue a trigger. Glut1 deficiency: fatigue-associated acute ataxia, quadriplegia, dystonia. |
|
Weissbach [159-161] |
Movement Disorders Society Gene Review: pathogenic variants in five genes associated with DRD: aside from those mentioned above, QDPR Most common DYT/PARK-GCH1 variant is a single nucleotide variant leading to a premature stop codon in exon 1 Discussion of genetic (monogenic) atypical PD and DRD. Red flags for atypical PD: supranuclear gaze palsy in ATP13A2 and hypoventilation in mutations of the DCTN1 gene (Perry syndrome: described in a Canadian family, AD, adult-onset parkinsonism, sleep problems and respiratory trouble; may have PSP phenotype) |
|
Li [162] |
Case report in “DYT24”-ANO3, a “Chinese dystonia”: 12-year-old female with left leg abnormal postures since age 11 which improved with sleep; then a progressive gait disturbance with eventual response to DBS. |
|
Abbreviations used: ADCY5: Adenylate Cyclase 5; ANO3: Anoctamin 3; ATP1A3: Adenosine Triphosphate Synthase, Sodium/Potassium Transporting, Alpha-3 Polypeptide; ATP13A2: Adenosine Triphosphate Synthase, Sodium/Potassium Transporting, Alpha-2 Polypeptide; BH4: Tetrahydrobiopterin; DCTN1: Dynactin; DDC: Dopa Decarboxylase; DHPR: Dihydropteridine Reductase; DnaJC12: DnaJ Heat Shock Protein Family Member C12; DRD: Dopa-Responsive Dystonia, Segawa Disease; DYT/PARK: Dystonia/Parkinsonism; GCH1: Guanosine Triphosphate (GTP) Cyclohydrolase 1; Glut1: Glucose Transporter 1; MxMD: Mixed Movement Disorder; PCD: Pterin-4a-Carbinolamine Dehydrase; PDE: Phosphodiesterase; PTP: 6-Pyruvoyl-Tetrahydropterin; QDPR: Quinoid Dihydropteridine Reductase; SLC6A3: Solute Carrier Family 6 Member A3 (encodes the dopamine transporter or DAT); SLC18A2: Solute Carrier Family 18 Member A2 (encodes vesicular monoamine transporter 2 or VMAT2); SPR: Sepiapterin Reductase; TH: Tyrosine Hydroxylase |
|
Why co-contraction in dystonia?
Pleiotropy is the rule rather than the exception in all the syndromes described in Table 5, but we concentrate on the phenomenon of co-contraction of agonist and antagonist muscles that results in a fixed posture in dystonia.
Muscles across joints achieve points of equilibrium in external space, based on coordinated activities of agonist and antagonist muscles in a limb. DA as well as other amines may directly influence neural modules in cord, as we discussed with specific reference to the A11 DA cell cluster.
Broadly tuned cortical neurons probably do not stipulate the specific brainstem or spinal cord modules to generate a combination of muscle forces—what has been termed a “motor field”—with nested equilibrium points [163]. To address the surfeit of potential effectors in a normal action, a motor task is more likely divided into well-timed subtasks associated with discrete neural substrates. Both subcortical (in the broadest sense of “subcortex,” even those in cord) and cortical modules may parse a large computational problem into parts [164].
Co-contraction of muscles that should not co-contract is a timing problem in miniature: agonists and antagonists should not contract simultaneously at any given moment, and co-contraction is powerfully inhibited by DA [165]. As many clinical presentations in DRD suggest, co-contraction can also vary across the time frame of a day. DA dysregulation is not an epiphenomenon of a clock disturbance: the dysregulation itself may result in mis-timing across momentary, short, and long intervals.
Conclusions Relevant to the Time Domain
Table 6 offers our major points and shortcomings in this paper.
Timing is a function of central and peripheral clocks whose interaction happens across a wide net; many lesion locales, including in dopaminergic nuclei, could disturb the percept of time. Albrecht [166] has quipped that, at least for mammals, there may be as many biological clocks as there are cells in the body.
Variable presentations in antibody-mediated syndromes and pleiotropy in genetic disease may set the stage for diagnostic uncertainty, but suggestions of a timing disturbance are hints, readily elicited by history or examination, that can aid in clinical accuracy.
Motor variation during a waking day happens both in PD and among normals. But, in PD, there is less of a difference between the most and least active epochs during the day. In RBD, absence of atonia predicts clinical Parkinsonism once alternative explanations have been excluded.
|
1. |
Movement disorders may vary with time of day and with mental state, including states of reduced consciousness. |
|
2. |
Despite hypothalamic control of biological timing, many cell populations, including dopaminergic nuclei, have “clock” functions. |
|
3. |
Parkinsonism and the synucleinopathies illustrate our argument, in light of several aspects: a behavioral disorder in sleep before the appearance of motor signs, diurnal variation in severity of motor manifestations, and nocturnal movements. |
|
4. |
A role for an inflammatory or autoimmune mechanism for timing or state variability in movement disorders becomes especially clear in various antibody-associated diseases. |
|
5. |
Major entities to consider relevant to point four include: anti-IgLON5, anti-NMDAR, anti-CASPR2, anti-LGI1, anti-Ma2, anti-DPPX, and anti-GABABR. |
|
6. |
Willis-Ekbom (restless legs syndrome) is an akathisia that implicates both iron and dopamine; clinically, it is a movement disorder associated with either recumbency or sleep (especially given the relationship between Willis Ekbom and stereotyped periodic leg movements of sleep or resting wakefulness). |
|
7. |
Dystonias specifically with diurnal variation are often monogenic, and we discuss them in relationship to the pathways of dopamine synthesis. |
|
Shortcomings |
|
|
1. |
Our methods for literature review were various, not consistent. |
|
2. |
Ours cannot be considered a systematic review, but rather simply a “primer.” |
|
3. |
We merely speculate on possible dopamine-related mechanisms of dystonia. |
|
4. |
We do not provide a neurological localization for timing disturbances and associated movement disorders. |
Is clock dysfunction evidence of a degenerative process?
It need not be, insofar as anti-IgLON5, anti-Ma2, anti-DPPX, and anti-LGI1 (just four examples) are not neurodegenerative in the same way as PD. But RBD is a red flag in those antibody syndromes as well as in Parkinsonism both before and after the advent of motor or cognitive signs or both.
Hyperkinesis in a state of reduced consciousness is not pathognomonic of any disease, but observing the hyperkinesis invites consideration of context, especially autoimmunity. By history or laboratory study, evidence of status dissociatus/agrypnia excitata should prompt suspicion of anti-NMDAR, anti-CASPR2, and anti-DPPX. Also, as Table 4 suggests, inflammatory disease and neurodegeneration are not mutually exclusive: immune mediation may contribute to neural dysfunction or frank cell loss in concert with a primary pathogenic process or in the absence of such a process.
The major implication of a temporal disturbance is that a lesion may result in a system disturbance, but the disturbance itself, not necessarily lesion localization, is the interest. Frank absence of a lesion, likewise, does not obviate a network problem, especially if key pathogenic variables are genetic and metabolic in nature, as in Table 5.
With respect to the hypothalamic A11 DA group, Earley et al. [167] compared six RLS and six age-matched control autopsy cases to find that there was no significant difference in A11 cells between groups. The authors conceded that A11 could still be involved in RLS pathogenesis; their caution is appropriate in light of evidence that DA deficiency can occur in the absence of DA neuronal loss, as has been described in DRD [168].
Similarly, in dystonias generally, neuropathological reports have been limited in their number and scope—e.g., a single report of nigral cell loss in a variant DRD [169]. A general caution still applies: absence or presence of a lesion neither obviates nor confirms a network dysfunction that can be ascertained clinically.
What is the relationship between putative cause and specific phenotype?
To say, “one genetic abnormality, multiple phenotypes,” as one might ascertain from Table 5, under-represents how multiple mutations can happen in a gene. To say, “multiple phenotypes, one disease,” as could be said of antibody-mediated syndromes, probably reveals how little we understand about whether (or if) production of an antibody causes a network disturbance—and if it does, the mechanism by which a time-of-day manifestations occur is not therefore obvious.
In many neurodegenerations, observed pathology may or may not relate to a specific network disturbance in the time domain. Alpha-synuclein aggregates in various non-brain, peripheral locales, including salivary glands, colon, and skin [170]. In AD, with its stereotyped and staged progression in the brain neuropathologically, RBD is rare at all stages of the disease. Likewise, RBD is not a phenotype in HD. Nevertheless, RLS and PLMS are present in synucleinopathy, tauopathy, and some trinucleotide-repeat diseases. Many lesion locales can result in network derangement [171], perhaps across the breadth of the neuraxis, so that specific localizations are not possible.
The relationship between etiology and phenotype begs a question that arises about pleiotropy in genetics [172]: if pleiotropy refers to “different kinds of one-to-many maps,” then what map is in question? Clinical phenomenology in the time domain or in relationship to mental state is an underexplored landscape in the study of movement disorders.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author Contributions
Acquisition, analysis, interpretation of data; drafting of work, revision for content; approval of publication; accountability for all aspects of the work: J. S-O., S.C., M.C., E.M.
Funding
None.
Supplementary Material
Supplementary Material is available in the on-line version of this paper.
Data Availability Statement
All relevant data are contained within the article. The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
References
2. Miyawaki EK. Review: Subjective Time Perception, Dopamine Signaling, and Parkinsonian Slowness. Front Neurol. 2022 Jul 8;13:927160.
3. Walker WH, Liu JA, Nelson RJ. Disruptions of Circadian Rhythms and Sleep/Wake Cycles in Neurologic Disorders. Sleep and Clocks in Aging and Longevity. 2023 Mar 4:461-80.
4. Cullell N, Cárcel-Márquez J, Gallego-Fábrega C, Muiño E, Llucià-Carol L, Lledós M, et al. Sleep/wake cycle alterations as a cause of neurodegenerative diseases: A Mendelian randomization study. Neurobiol Aging. 2021 Oct;106:320.e1-320.e12.
5. Gros P, Videnovic A. Overview of Sleep and Circadian Rhythm Disorders in Parkinson Disease. Clin Geriatr Med. 2020 Feb;36(1):119-30.
6. Hood S, Amir S. Neurodegeneration and the Circadian Clock. Front Aging Neurosci. 2017 May 30;9:170.
7. Smilowska K, van Wamelen DJ, Bloem BR. The multimodal effect of circadian interventions in Parkinson's disease: A narrative review. Parkinsonism Relat Disord. 2023 May;110:105309.
8. Gaig C, Graus F. Motor symptoms in nonparaneoplastic CNS disorders associated with neural antibodies. Handb Clin Neurol. 2023;196:277-294.
9. Ferré S, Guitart X, Quiroz C, Rea W, García-Malo C, Garcia-Borreguero D, et al. Akathisia and Restless Legs Syndrome: Solving the Dopaminergic Paradox. Sleep Med Clin. 2021 Jun;16(2):249-67.
10. Stephen CD. The Dystonias. Continuum (Minneap Minn). 2022 Oct 1;28(5):1435-75.
11. Allada R, Bass J. Circadian Mechanisms in Medicine. N Engl J Med. 2021 Feb 11;384(6):550-61.
12. Paul JR, Davis JA, Goode LK, Becker BK, Fusilier A, Meador-Woodruff A, Gamble KL. Circadian regulation of membrane physiology in neural oscillators throughout the brain. Eur J Neurosci. 2020 Jan;51(1):109-38.
13. Leng Y, Musiek ES, Hu K, Cappuccio FP, Yaffe K. Association between circadian rhythms and neurodegenerative diseases. Lancet Neurol. 2019 Mar;18(3):307-18.
14. Abe M, Herzog ED, Yamazaki S, Straume M, Tei H, Sakaki Y, et al. Circadian rhythms in isolated brain regions. J Neurosci. 2002 Jan 1;22(1):350-6.
15. Hood S, Amir S. The aging clock: circadian rhythms and later life. J Clin Invest. 2017 Feb 1;127(2):437-46.
16. Volicer L, Harper DG, Manning BC, Goldstein R, Satlin A. Sundowning and circadian rhythms in Alzheimer's disease. Am J Psychiatry. 2001 May;158(5):704-11.
17. Gnanasekaran G. "Sundowning" as a biological phenomenon: current understandings and future directions: an update. Aging Clin Exp Res. 2016 Jun;28(3):383-92.
18. Postuma RB, Iranzo A, Hu M, Högl B, Boeve BF, Manni R, et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: a multicentre study. Brain. 2019 Mar 1;142(3):744-59.
19. Oz S, Dagay A, Katzav S, Wasserman D, Tauman R, Gerston A, et al. Monitoring sleep stages with a soft electrode array: Comparison against vPSG and home-based detection of REM sleep without atonia. J Sleep Res. 2023 Oct;32(5):e13909.
20. Porter VR, Avidan AY. Clinical Overview of REM Sleep Behavior Disorder. Semin Neurol. 2017 Aug;37(4):461-70.
21. Antelmi E, Lippolis M, Biscarini F, Tinazzi M, Plazzi G. REM sleep behavior disorder: Mimics and variants. Sleep Med Rev. 2021 Dec;60:101515.
22. Guttowski D, Mayer G, Oertel WH, Kesper K, Rosenberg T. Validation of semiautomatic scoring of REM sleep without atonia in patients with RBD. Sleep Med. 2018 Jun;46:107-13.
23. Arnulf I. REM sleep behavior disorder: motor manifestations and pathophysiology. Mov Disord. 2012 May;27(6):677-89.
24. Figorilli M, Meloni M, Lanza G, Casaglia E, Lecca R, Saibene FL, et al. Considering REM Sleep Behavior Disorder in the Management of Parkinson's Disease. Nat Sci Sleep. 2023 May 5;15:333-52.
25. van Wamelen DJ, Urso D, Ray Chaudhuri K. How Time Rules: Diurnal Motor Patterns in de novo Parkinson's Disease. J Parkinsons Dis. 2021;11(2):695-702.
26. Stewart J, Bachman G, Cooper C, Liu L, Ancoli-Israel S, Alibiglou L. Circadian dysfunction and fluctuations in gait initiation impairment in Parkinson's disease. Exp Brain Res. 2018 Mar;236(3):655-64.
27. Mirelman A, Volkov J, Salomon A, Gazit E, Nieuwboer A, Rochester L, et al. Digital Mobility Measures: A Window into Real-World Severity and Progression of Parkinson's Disease. Mov Disord. 2024 Feb;39(2):328-38.
28. Marano M, Rosati J, Magliozzi A, Casamassa A, Rappa A, Sergi G, et al. Circadian profile, daytime activity, and the Parkinson's phenotype: A motion sensor pilot study with neurobiological underpinnings. Neurobiol Sleep Circadian Rhythms. 2023 Mar 26;14:100094.
29. Moreau C, Rouaud T, Grabli D, Benatru I, Remy P, Marques AR, et al. Overview on wearable sensors for the management of Parkinson's disease. NPJ Parkinsons Dis. 2023 Nov 2;9(1):153.
30. Fifel K, Videnovic A. Circadian alterations in patients with neurodegenerative diseases: Neuropathological basis of underlying network mechanisms. Neurobiol Dis. 2020 Oct;144:105029.
31. Videnovic A, Golombek D. Circadian Dysregulation in Parkinson's Disease. Neurobiol Sleep Circadian Rhythms. 2017 Jan;2:53-8.
32. Videnovic A, Willis GL. Circadian system - A novel diagnostic and therapeutic target in Parkinson's disease? Mov Disord. 2016 Mar;31(3):260-9.
33. Suzuki K. Current Update on Clinically Relevant Sleep Issues in Parkinson's Disease: A Narrative Review. J Parkinsons Dis. 2021;11(3):971-92.
34. Mirelman A, Hillel I, Rochester L, Del Din S, Bloem BR, Avanzino L, et al. Tossing and Turning in Bed: Nocturnal Movements in Parkinson's Disease. Mov Disord. 2020 Jun;35(6):959-68.
35. Högl B, Arnulf I, Comella C, Ferreira J, Iranzo A, Tilley B, et al. Scales to assess sleep impairment in Parkinson's disease: critique and recommendations. Mov Disord. 2010 Dec 15;25(16):2704-16.
36. Kurtis MM, Balestrino R, Rodriguez-Blazquez C, Forjaz MJ, Martinez-Martin P. A Review of Scales to Evaluate Sleep Disturbances in Movement Disorders. Front Neurol. 2018 May 29;9:369.
37. Zhu XY, Liu Y, Zhang XJ, Yang WH, Feng Y, Ondo WG, et al. Clinical characteristics of leg restlessness in Parkinson's disease compared with idiopathic Restless Legs Syndrome. J Neurol Sci. 2015 Oct 15;357(1-2):109-14.
38. Matsubara T, Suzuki K, Fujita H, Watanabe Y, Sakuramoto H, Matsubara M, et al. Restless legs syndrome, leg motor restlessness and their variants in patients with Parkinson's disease and related disorders. J Neurol Sci. 2018 Oct 15;393:51-7.
39. Krishnan PR, Bhatia M, Behari M. Restless legs syndrome in Parkinson's disease: a case-controlled study. Mov Disord. 2003 Feb;18(2):181-5.
40. Nomura T, Inoue Y, Miyake M, Yasui K, Nakashima K. Prevalence and clinical characteristics of restless legs syndrome in Japanese patients with Parkinson's disease. Mov Disord. 2006 Mar;21(3):380-4.
41. Shin HY, Youn J, Yoon WT, Kim JS, Cho JW. Restless legs syndrome in Korean patients with drug-naïve Parkinson's disease: a nation-wide study. Parkinsonism Relat Disord. 2013 Mar;19(3):355-8.
42. Kim JM, Choi SM, Cho SH, Kim BC. Restless legs syndrome affects sleep in de novo Parkinson's disease patients. Medicine (Baltimore). 2023 Nov 3;102(44):e35551.
43. Högl B, Stefani A. Restless legs syndrome and periodic leg movements in patients with movement disorders: Specific considerations. Mov Disord. 2017 May;32(5):669-81.
44. De Cock VC, Lannuzel A, Verhaeghe S, Roze E, Ruberg M, Derenne JP, et al. REM sleep behavior disorder in patients with guadeloupean parkinsonism, a tauopathy. Sleep. 2007 Aug;30(8):1026-32.
45. Nollet M, Franks NP, Wisden W. Understanding Sleep Regulation in Normal and Pathological Conditions, and Why It Matters. J Huntingtons Dis. 2023;12(2):105-19.
46. French IT, Muthusamy KA. A Review of Sleep and Its Disorders in Patients with Parkinson's Disease in Relation to Various Brain Structures. Front Aging Neurosci. 2016 May 23;8:114.
47. Li S, Wang Y, Wang F, Hu LF, Liu CF. A New Perspective for Parkinson's Disease: Circadian Rhythm. Neurosci Bull. 2017 Feb;33(1):62-72.
48. Hasegawa E, Miyasaka A, Sakurai K, Cherasse Y, Li Y, Sakurai T. Rapid eye movement sleep is initiated by basolateral amygdala dopamine signaling in mice. Science. 2022 Mar 4;375(6584):994-1000.
49. Mariotti P, Quaranta D, Di Giacopo R, Bentivoglio AR, Mazza M, Martini A, et al. Rapid eye movement sleep behavior disorder: a window on the emotional world of Parkinson disease. Sleep. 2015 Feb 1;38(2):287-94.
50. Rathor P, Ch R. Metabolic Basis of Circadian Dysfunction in Parkinson's Disease. Biology (Basel). 2023 Sep 28;12(10):1294.
51. Fonken LK, Frank MG, Kitt MM, Barrientos RM, Watkins LR, Maier SF. Microglia inflammatory responses are controlled by an intrinsic circadian clock. Brain Behav Immun. 2015 Mar;45:171-9.
52. Liu WW, Wei SZ, Huang GD, Liu LB, Gu C, Shen Y, et al. BMAL1 regulation of microglia-mediated neuroinflammation in MPTP-induced Parkinson's disease mouse model. FASEB J. 2020 May;34(5):6570-81.
53. Trotti LM. Central Disorders of Hypersomnolence. Continuum (Minneap Minn). 2020 Aug;26(4):890-907.
54. Balint B, Vincent A, Meinck HM, Irani SR, Bhatia KP. Movement disorders with neuronal antibodies: syndromic approach, genetic parallels and pathophysiology. Brain. 2018 Jan 1;141(1):13-36.
55. Gövert F, Leypoldt F, Junker R, Wandinger KP, Deuschl G, Bhatia KP, et al. Antibody-related movement disorders - a comprehensive review of phenotype-autoantibody correlations and a guide to testing. Neurol Res Pract. 2020 Feb 20;2:6.
56. Kubick N, Brösamle D, Mickael ME. Molecular Evolution and Functional Divergence of the IgLON Family. Evol Bioinform Online. 2018 May 21;14:117693431877508.
57. Sabater L, Gaig C, Gelpi E, Bataller L, Lewerenz J, Torres-Vega E, et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol. 2014 Jun;13(6):575-86.
58. Gaig C, Graus F, Compta Y, Hoegl B, Bataller L, Brueggemann N, et al. Clinical manifestations of the anti-IgLON5 disease. Neurology. 2017 May 2;88(18):1736-43.
59. Honorat JA, Komorowski L, Josephs KA, Fechner K, St Louis EK, Hinson SR, et al. IgLON5 antibody: Neurological accompaniments and outcomes in 20 patients. Neurol Neuroimmunol Neuroinflamm. 2017 Jul 18;4(5):e385.
60. Gaig C, Compta Y, Heidbreder A, Marti MJ, Titulaer MJ, Crijnen Y, et al. Frequency and characterization of movement disorders in anti-IgLON5 disease. Neurology. 2021 Oct 4;97(14):e1367-81.
61. Atadzhanov M, Chishimba L. Recognition of Movement Disorders as Cardinal Features of Anti-IgLON5 Disease: Expanding the Clinical Spectrum. Neurology. 2021 Oct 4;97(14):661-2.
62. Ni Y, Shen D, Zhang Y, Song Y, Gao Y, Zhou Q, et al. Expanding the clinical spectrum of anti-IgLON5 disease: a multicenter retrospective study. Eur J Neurol. 2022 Jan;29(1):267-76.
63. Grueter T, Moellers FE, Tietz A, Dargvainiene J, Melzer N, Heidbreder A, et al. Clinical, serological and genetic predictors of response to immunotherapy in anti-IgLON5 disease. Brain. 2023 Feb 13;146(2):600-611.
64. Madetko N, Marzec W, Kowalska A, Przewodowska D, Alster P, Koziorowski D. Anti-IgLON5 disease—the current state of knowledge and further perspectives. Front Immunol. 2022 Mar 1;13:852215.
65. Graus F, Vogrig A, Muniz-Castrillo S, Antoine J-CG, Desestret V, Dubey D, et al. Updated diagnostic criteria for paraneoplastic neurologic syndromes. Neurol Neuroimmunol Neuroinflamm. 2021 May 18;8(4):e1014.
66. Heidbreder A, Philipp K. Anti-IgLON 5 Disease. Curr Treat Options Neurol. 2018 Jun 23;20(8):29.
67. Pi Y, Zhang LL, Li JC. Anti-IgLON5 disease with distinctive brain MRI findings responding to immunotherapy: A case report. Medicine (Baltimore). 2021 Jan 29;100(4):e24384.
68. Chen H, Wu J, Irani SR. Distinctive Magnetic Resonance Imaging Findings in IgLON5 Antibody Disease. JAMA Neurol. 2020 Jan 1;77(1):125-26.
69. Tagliapietra M, Frasson E, Cardellini D, Mariotto S, Ferrari S, Zanusso G, et al. Hypothalamic-bulbar MRI hyperintensity in anti-IgLON5 disease with serum-restricted antibodies: a case report and systematic review of literature. J Alzheimers Dis. 2021;79(2):683-91.
70. Blinder T, Lewerenz J. Cerebrospinal Fluid Findings in Patients With Autoimmune Encephalitis-A Systematic Analysis. Front Neurol. 2019 Jul 25;10:804.
71. Giannoccaro MP, Verde F, Morelli L, Rizzo G, Ricciardiello F, Liguori R. Neural surface antibodies and neurodegeneration: clinical commonalities and pathophysiological relationships. Biomedicines. 2023 Feb 22;11(3):666.
72. Dalmau J, Armangué T, Planagumà J, Radosevic M, Mannara F, Leypoldt F, et al. An update on anti-NMDA receptor encephalitis for neurologists and psychiatrists: mechanisms and models. Lancet Neurol. 2019 Nov;18(11):1045-57.
73. Gable MS, Sheriff H, Dalmau J, Tilley DH, Glaser CA. The frequency of autoimmune N-methyl-D-aspartate receptor encephalitis surpasses that of individual viral etiologies in young individuals enrolled in the California Encephalitis Project. Clin Infect Dis. 2012 Apr;54(7):899-904.
74. Vitaliani R, Mason W, Ances B, Zwerdling T, Jiang Z, Dalmau J. Paraneoplastic encephalitis, psychiatric symptoms, and hypoventilation in ovarian teratoma. Ann Neurol. 2005 Oct;58(4):594-604.
75. Armangué T, Leypoldt F, Málaga I, Raspall-Chaure M, Marti I, Nichter C, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol. 2014 Feb;75(2):317-23.
76. Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008 Dec;7(12):1091-8.
77. Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011 Jan;10(1):63-74.
78. Maneta E, Garcia G. Psychiatric manifestations of anti-NMDA receptor encephalitis: neurobiological underpinnings and differential diagnostic implications. Psychosomatics. 2014 Jan-Feb;55(1):37-44.
79. Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013 Feb;12(2):157-65.
80. Blattner MS, Day GS. Sleep disturbances in patients with autoimmune encephalitis. Curr Neurol Neurosci Rep. 2020 Jun 10;20(7):28.
81. Stamelou M, Plazzi G, Lugaresi E, Edwards MJ, Bhatia KP. The distinct movement disorder in anti-NMDA receptor encephalitis may be related to status dissociatus: a hypothesis. Mov Disord. 2012 Sep 15;27(11):1360-3.
82. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016 Apr;15(4):391-404.
83. Irani SR, Bera K, Waters P, Zuliani L, Maxwell S, Zandi MS, et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain. 2010 Jun;133(Pt 6):1655-67.
84. Wang R, Guan H-Z, Ren H-T, Wang W, Hong Z, Zhou D. CSF findings in patients with anti-N-methyl-d-aspartate receptor-encephalitis. Seizure. 2015 Jul;29:137-42.
85. Iizuka T, Kaneko J, Tominaga N, Someko H, Nakamura M, Ishima D, et al. Association of progressive cerebellar atrophy with long-term outcome in patients with anti-N-methyl-d-aspartate receptor encephalitis. JAMA Neurol. 2016 Jun 1;73(6):706-13.
86. Florance NR, Davis RL, Lam C, Szperka C, Zhou L, Ahmad S, et al. Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis in children and adolescents. Ann Neurol. 2009 Jul;66(1):11-8.
87. Zrzavy T, Endmayr V, Bauer J, Macher S, Mossaheb N, Schwaiger C, et al. Neuropathological variability within a spectrum of NMDAR-encephalitis. Ann Neurol. 2021 Nov;90(5):725-37.
88. Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuilani L, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain. 2010 Sep;133(9):2734-48.
89. Van Sonderen A, Ariño H, Petit-Pedrol M, Leypoldt F, Koertvélyessy P, Wandinger KP, et al. The clinical spectrum of Caspr2 antibody-associated disease. Neurology. 2016 Aug 2;87(5):521-8.
90. Lugaresi E, Provini F. Agrypnia excitata: clinical features and pathophysiological implications. Sleep Med Rev. 2001 Aug;5(4):313-22.
91. Montagna P, Lugaresi E. Agrypnia excitata: a generalized overactivity syndrome and a useful concept in the neurophysiology of sleep. Clin Neurophysiol. 2002 Apr;113(4):552-60.
92. Cornelius JR, Pittock SJ, McKeon A, Lennon VA, Aston PA, Josephs KA, et al. Sleep manifestations of voltage-gated potassium channel complex autoimmunity. Arch Neurol. 2011 Jun;68(6):733-8.
93. Liguori R, Vincent A, Clover L, Avoni P, Plazzi G, Cortelli P, et al. Morvan’s syndrome: peripheral and central nervous system and cardiac involvement with antibodies to voltage-gated potassium channels. Brain. 2001 Dec;124(Pt 12):2417-26.
94. Irani SR, Pettingill P, Kleopa KA, Schiza N, Waters P, Mazia C, et al., Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol. 2012 Aug;72(2):241-55.
95. Qin X, Yang H, Zhu F, Wang Q, Shan W. Clinical character of CASPR2 autoimmune encephalitis: a multiple center retrospective study. Front Immunol. 2021 May 13;12:652864.
96. Shivaram S, Nagappa M, Seshagiri DV, Mahadevan A, Gangadhar Y, Sathyaprabha TN, et al. Clinical profile and treatment response in patients with CASPR2 antibody-associated neurological disease. Ann Indian Acad Neurol. 2021 Mar-Apr;24(2):178-85.
97. Tan KM, Lennon VA, Klein CJ, Boeve BF, Pittock SJ. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology. 2008 May 13;70(20):1883-90.
98. Duerr M, Nissen G, Suehs KW, Schwenkenbecher P, Geis C, Ringelstein M, et al.; German Network for Research on Autoimmune Encephalitis. CSF findings in acute NMDAR and LGI1 antibody-associated autoimmune encephalitis. Neurol Neuroimmunol Neuroinflamm. 2021 Oct 25;8(6):e1086.
99. Thompson J, Bi M, Murchison AG, Makuch M, Bien CG, Chu K, et al. The importance of early immunotherapy in patients with faciobrachial dystonic seizures. Brain. 2018 Feb 1;141(2):348-56.
100. Irani SR, Michell AW, Lang B, Pettingill P, Waters P, Johnson MR, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol. 2011 May;69(5):892-900.
101. Nantes JC, Thomas AG, Voets NL, Best JG, Rosenthal CR, Al-Diwani A, et al. Hippocampal functional dynamics are clinically implicated in autoimmune encephalitis with faciobrachial dystonic seizures. Front Neurol. 2018 Sep 4;9:736.
102. Striano P. Faciobrachial dystonic attacks: seizures or movement disorder? [author reply Irani SR, Michell AW, Johnson MR, et al.] Ann Neurol. 2011 Jul;70(1):179-80.
103. Irani SR, Stagg CJ, Schott JM, Rosenthal CR, Schneider SA, Pettingill P, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain. 2013 Oct;136(Pt 10):3151-62.
104. Graus F, Delattre JY, Antoine JC, Dalmau J, Giometto B, Grisold W, et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry. 2004 Aug;75(8):1135-40.
105. Hoffmann LA, Jarius S, Pellkofer HL, Schueller M, Krumbholz M, Koenig F, et al. Anti-Ma and anti-Ta associated paraneoplastic neurological syndromes: 22 newly diagnosed patients and review of previous cases. J Neurol Neurosurg Psychiatry. 2008 Jul;79(7):767-73.
106. Voltz R, Gultekin SH, Rosenfeld MR, Gerstner E, Eichen J, Posner JB, et al. A serologic marker of paraneoplastic limbic and brain-stem encephalitis in patients with testicular cancer. N Engl J Med. 1999 Jun 10;340(23):1788-95.
107. Rosenfeld MR, Eichen JG, Wade DF, Posner JB, Dalmau J. Molecular and clinical diversity in paraneoplastic immunity to Ma proteins. Ann Neurol. 2001 Sep;50(3):339-48.
108. Dalmau J, Graus F, Villarejo A, Posner JB, Blumenthal D, Thiessen B, et al. Clinical analysis of anti-Ma2-associated encephalitis. Brain. 2004 Aug;127(Pt 8):1831-44.
109. Devine MF, St Louis EK. Sleep disturbances associated with neurological autoimmunity. Neurotherapeutics. 2021 Jan;18(1):181-201.
110. Dauvilliers Y, Bauer J, Rigau V, Lalloyer N, Labauge P, Carlander B, et al. Hypothalamic immunopathology in anti-Ma–associated diencephalitis with narcolepsy-cataplexy. JAMA Neurol. 2013 Oct;70(10):1305-10.
111. Tyree SM, Borniger JC, de Lecea L. Hypocretin as a hub for arousal and motivation. Front Neurol. 2018 Jun 6;9:413.
112. Boronat A, Gelfand JM, Gresa-Arribas N, Jeong H-Y, Walsh M, Roberts K, et al. Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann Neurol. 2013 Jan;73(1):120-8.
113. Oliver Tobin W, Lennon VA, Komorowski L, Probst C, Clardy SL, Aksamit AJ, et al. DPPX potassium channel antibody: frequency, clinical accompaniments, and outcomes in 20 patients. Neurology. 2014 Nov 11;83(20):1797-803.
114. Gadoth A, Devine MF, Pittock SJ, McKeon A, Oliver Tobin W, Gossard TR, et al. Sleep disturbances associated with DPPX autoantibodies: a case series. J Neurol. 2023 Jul;270(7):3543-52.
115. Hara M, Ariño H, Petit-Pedrol M, Sabater L, Titulaer MJ, Martinez-Hernandez E, et al. DPPX antibody–associated encephalitis: main syndrome and antibody effects. Neurology. 2017 Apr 4;88(14):1340-8.
116. Batocchi AP, Della Marca G, Mirabella M, Caggiula M, Frisullo G, Mennuni GF, et al. Relapsing-remitting autoimmune agrypnia. Ann Neurol. 2001 Nov;50(5):668-71.
117. Frisullo G, Della Marca G, Mirabella M, Caggiula M, Broccolini A, Rubino M, et al. A human anti-neural autoantibody against GABA B receptor induces experimental autoimmune agrypnia. Exp Neurol. 2007 Apr;204(2):808-18.
118. Zhu F, Shan W, Lv R, Li Z, Wang Q. Clinical characteristics of anti-GABA-B receptor encephalitis. Front Neurol. 2020 May 21;11:403.
119. Guan HZ, Ren HT, Yang XZ, Lu Q, Peng B, Zhu YC, et al. Limbic encephalitis associated with anti-gamma-aminobutyric acid B receptor antibodies: a case series from China. Chin Med J (Engl). 2015 Nov 20;128(22):3023-8.
120. Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol. 2010 Jan;9(1):67-76.
121. Höftberger R, Titulaer MJ, Sabater L, Dome B, Rózsás A, Hegedus B, et al. Encephalitis and GABAB receptor antibodies: novel findings in a new case series of 20 patients. Neurology. 2013 Oct 22;81(17):1500-6.
122. Bonorat A, Sabater L, Saiz A, Dalmau J, Graus F. GABA(B) receptor antibodies in limbic encephalitis and anti-GAD-associated neurologic disorders. Neurology. 2011 Mar 1;76(9):795-800.
123. Chung HY, Wickel J, Voss A, Ceanga M, Sell J, Witte OW, et al. Autoimmune encephalitis with anti-IgLON5 and anti-GABAB-receptor antibodies: a case report. Medicine (Baltimore). 2019 May;98(20):e15706.
124. McKay J H, Dimberg EL, Chiriboga ASL. A systematic review of gamma-aminobutyric acid receptor type B autoimmunity. Neurol Neurochir Pol. 2019;53(1):1-7.
125. Budhram A, Mills JR, Shouman K, Dyck PJB, Hassan A, Zalewski NL. False-positive anti-neuronal nuclear antibody type 1 in a patient with RFC1 repeat expansion: preventing “phenotypic creep” in autoimmune neurology. J Neurol Sci. 2020 Sep 15;416:117018.
126. Gresa-Arribas N, Planagumà J, Petit-Pedrol M, Kawachi I, Katada S, Glaser CA, et al. Human neurexin-3alpha antibodies associate with encephalitis and alter synapse development. Neurology. 2016 Jun 14;86(24):2235-42.
127. Loehrer PA, Bien CI, Dusoi A-E, Timmerman L, Simon OJ. Neurexin-3alpha-associated autoimmune encephalitis: a case report of full recovery after rituximab therapy. Eur J Neurol. 2020 Dec;27(12):e91-3.
128. Mandel-Brehm C, Benson LA, Tran B, Kung AF, Mann SA, Vazquez SE, et al. ZSCAN1 autoantibodies are associated with pediatric paraneoplastic ROHHAD. Ann Neurol. 2022 Aug;92(2):279-91.
129. Dale RC, Church AJ, Cardoso F, Goddard E, Cox TC, Chong WK, et al. Poststreptococcal acute disseminated encephalomyelitis with basal ganglia involvement and auto-reactive antibasal ganglia antibodies. Ann Neurol. 2001 Nov;50(5):588-95.
130. Dale RC, Irani SR, Brilot F, Pillai S, Webster R, Gill D, et al. N-methyl-D-aspartate receptor antibodies in pediatric dyskinetic encephalitis lethargica. Ann Neurol. 2009 Nov;66(5):704-9.
131. Dale RC, Merheb V, Pillai S, Wang D, Cantrill L, Murphy TK, et al. Antibodies to surface dopamine-2 receptor in autoimmune movement and psychiatric disorders. Brain. 2012 Nov;135(Pt 11):3453-68.
132. Pathmanandavel K, Starling J, Merheb V, Ramanathan S, Sinmaz N, Dale RC, et al. Antibodies to surface dopamine-2 receptor and N-methyl-D-aspartate receptor in the first episode of acute psychosis in children. Biol Psychiatry. 2015 Mar 15;77(6):537-47.
133. Mohammad SS, Sinclair K, Pillai S, Merheb V, Aumann TD, Gill D, et al. Herpes simplex encephalitis relapse with chorea is associated with autoantibodies to N-methyl-D-aspartate receptor or dopamine-2 receptor. Mov Disord. 2014 Jan;29(1):117-22.
134. Allen RP, Picchietti DL, Garcia-Borreguero D, Ondo WG, Walters AS, Winkelman JW, et al.; International Restless Legs Syndrome Study Group. Restless legs syndrome/Willis-Ekbom disease diagnostic criteria: updated International Restless Legs Syndrome Study Group (IRLSSG) consensus criteria—history, rationale, description, and significance. Sleep Med. 2014 Aug;15(8):860-73.
135. Aurora RN, Kristo DA, Bista SR, Rowley JA, Zak RS, Casey KR, et al.; American Academy of Sleep Medicine. The treatment of restless legs syndrome and periodic limb movement disorder in adults—an update for 2012: practice parameter with an evidence-based systematic review and meta-analyses: an American Academy of Sleep Medicine Clinical Practice Guideline. Sleep. 2012 Aug 1;35(8):1039-62.
136. Manconi M, Garcia-Borreguero D, Schormair B, Videnovic A, Berger K, Ferri R, Dauvilliers Y. Restless legs syndrome. Nature reviews Disease primers. 2021 Nov 3;7(1):80.
137. Earley C, Uhl GR, Clemens S, Ferré S. Connectomy and molecular pharmacological dopaminergic differences in RLS: plastic changes and neuroadaptations that may contribute to augmentation. Sleep Med. 2017 Mar;31:71-77.
138. Mackie S and Winkelman JW. Long-term treatment of Restless Legs Syndrome (RLS): an approach to management of worsening symptoms, loss of efficacy, and augmentation. CNS Drugs. 2015 May;29(5):351-7.
139. Berke JD. What does dopamine mean? Nat Neurosci. 2018 Jun;21(6):787-93.
140. Eban-Rothschild A, Rothschild G, Giardino WJ, Jones JR, de Lecea L. VTA dopaminergic neurons regulate ethologically relevant sleep-wake behaviors. Nat Neurosci. 2016 Oct;19(10):1356-66.
141. Ferris MJ, España RA, Locke JL, Konstantopoulos JK, Rose JH, Chen R,et al. Dopamine transporters govern diurnal variation in dopamine tone. Proc Natl Acad Sci U S A. 2014 Jul 1;111(26):E2751-9.
142. Olejniczak I, Begemann K, Wilhelm I, Oster H. The circadian neurobiology of reward. Acta Physiol (Oxf). 2023 Mar;237(3):e13928.
143. Allen RP. Restless leg syndrome/Willis-Ekbom disease pathophysiology. Sleep Med Clin. 2015 Sep;10(3):207-14.
144. Björklund A, Dunnett SB. Dopamine neuron systems in the brain: an update. Trends Neurosci. 2007 May;30(5):194-202.
145. Reinig S, Driever W, Arrenberg AB. The descending diencephalic dopamine system is tuned to sensory stimuli. Curr Biol. 2017 Feb 6;27(3):318-33.
146. Sharples SA, Koblinger K, Humphreys JM, Whelan PJ. Dopamine: a parallel pathway for the modulation of spinal locomotor networks. Front Neural Circuits. 2014 Jun 16;8:55.
147. Clemens S, Rye D, Hochman S. Restless legs syndrome: revisiting the dopamine hypothesis from the spinal cord perspective. Neurology. 2006 Jul 11;67(1):125-30.
148. Paulus W, Schomburg ED. Dopamine and the spinal cord in restless legs syndrome: does spinal cord physiology reveal a basis for augmentation? Sleep Med Rev. 2006 Jun;10(3):185-96.
149. Ferré S, Garcia Borreguero D, Allen RP, Earley CJ. New insights into the neurobiology of restless legs syndrome. Neuroscientist. 2019 Apr;25(2):113-25.
150. Segawa M, Hosaka A, Miyagawa F, Nomura Y, Imai H. Hereditary progressive dystonia with marked diurnal variation. In: Advances in Neurology, vol. 14, eds. R. Eldridge, S. Fahn. New York: Raven Press, p. 215-33.
151. Lee W-W, Jeon B, Kim R. Expanding the spectrum of dopa-responsive dystonia (DRD) and proposal for new definition: DRD, DRD-plus, and DRD look-alike. J Korean Med Sci. 2018 May 24;33(28):e184.
152. Salles PA, Terán-Jimenez M, Vidal-Santoro A, Chaná-Cuevas P, Kauffman M, Espay AJ. Recognizing atypical dopa-responsive dystonia and its mimics. Neurol Clin Pract. 2021 Dec;11(6):e876-84.
153. Stephen CD, Dy-Hollins M, Melo De Gusmao C, Al Qahtani X, Sharma N. Dystonias: clinical recognition and the role of additional diagnostic testing. Semin Neurol. 2023 Feb;43(1):17-34.
154. De Pablo-Fernandez E, Warner TT. Dystonia. Br Med Bull. 2017 Sep 1;123(1):91-102.
155. Keller Sarmiento IJ, Mencacci NE. Genetic dystonias: update on classification and new genetic discoveries. Curr Neurol Neurosci Rep. 2021 Feb 9;21(3):8.
156. Wong RSH, Mohammad S, Parayil Sankaran B, Junek R, Kim WT, Wotton T, et al. Developmental delay and non-phenylketonuria (PKU) hyperphenylalaninemia in DNAJC12 deficiency: case and approach. Brain Dev. 2023 Oct;45(9):523-31.
157. Carecchio M, Mencacci NE. Emerging monogenic complex hyperkinetic disorders. Curr Neurol Neurosci Rep. 2017 Oct 30;17(12):97.
158. Pérez-Dueñas B, Gorman K, Marcé-Grau A, Ortigoza-Escobar JD, Macaya A, Danti FR, et al. The genetic landscape of complex childhood-onset hyperkinetic movement disorders. Mov Disord. 2022 Nov;37(11):2197-209.
159. Weissbach A, Pauly MG, Herzog R, Hahn L, Halmans S, Hamami F, et al. Relationship of genotype, phenotype, and treatment in dopa-responsive dystonia: MDSGene review. Mov Disord. 2022 Feb;37(2):237-52.
160. Weissbach A, Saranza G, Domingo A. Combined dystonias: clinical and genetic updates. J Neural Transm (Vienna). 2021 Apr;128(4):417-29.
161. Weissbach A, Wittke C, Kasten M, Klein C. ‘Atypical’ Parkinson’s disease—genetic. Int Rev Neurobiol. 2019;149:207-235
162. Li S, Wang L, Yang Y, Ma J, Wan X. ANO3 mutations in Chinese dystonia: a genetic screening study using next-generation sequencing. Front Neurol. 2020 Feb 7;10:1351.
163. Poggio T, Bizzi E. Generalization in vision and motor control. Nature. 2004 Oct 14;431(7010):768-74.
164. Gershman SJ, Pesaran B, Daw ND. Human reinforcement learning subdivides structured action spaces by learning effector-specific values. J Neurosci. 2009 Oct 28;29(43):13524-31.
165. Schomburg ED, Steffens H. Comparative analysis of L-DOPA actions on nociceptive and non-nociceptive spinal reflex pathways in the cat. Neurosci Res. 1998 Aug;31(4):307-16.
166. Albrecht U. Timing to perfection: the biology of central and peripheral circadian clocks. Neuron. 2012 Apr 26;74(2):246-60.
167. Earley CJ, Allen RP, Connor JR, Ferrucci L, Troncoso J. The dopaminergic neurons of the A11 system in RLS autopsy brains appear normal. Sleep Med. 2009 Dec;10(10):1155-7.
168. Casper C, Kalliolia E, Warner TT. Recent advances in the molecular pathogenesis of dystonia-plus syndromes and heredodegenerative dystonias. Curr Neuropharmacol. 2013 Jan;11(1):30-40.
169. Sharma N. Neuropathology of dystonia. Tremor Other Hyperkinet Mov (N Y). 2019 Feb 25;9:569.
170. Wang Z, Becker K, Donadio V, Siedlak S, Yuan J, Rezaee M, et al. Skin alpha-synuclein aggregation seeding activity as a novel biomarker for Parkinson disease. JAMA Neurol. 2020 Sep 28;78(1):1-11.
171. Fifel K, Videnovic A. Circadian and sleep dysfunctions in neurodegenerative disorders—an update. Neurosci. 2021 Jan 18;14:627330
172. Paaby AB, Rockman MV. The many faces of pleiotropy. Trends Genet. 2013 Feb;29(2):66-73.