Abstract
Percutaneous transluminal coronary angioplasty (PTCA) with or without vascular stenting is commonly used to treat coronary heart disease. However, restenosis occurs in 30-50% of patients undergoing simple balloon angioplasty and in 10-30% of patients who receive an intravascular stent. Collagen exposure at the vascular damage site leads to platelet adhesion and aggregation mainly via the activation of glycoprotein (GP) VI (a primary platelet collagen receptor), which contributes to the generation of thromboxane (TX) A2 . It is produced from arachidonic acid by the activity of cyclooxygenase (COX)-1. TXA2 is a potent mediator of platelet aggregation, vasoconstriction, and vascular smooth muscle cells (VSMC) proliferation and migration. In patients undergoing PTCA, an early increase of the systemic biosynthesis of TXA2 occurs and is largely suppressed by low-dose Aspirin, a selective inhibitor of platelet COX-1. Thus, platelet activation and enhanced TXA2 biosynthesis in response to vascular damage and collagen exposure is an early event which may contribute to restenosis. In this review, we report and discuss the evidence obtained in vivo and in vitro that the blockage of the interaction of platelet GPVI with injured vascular components, using novel antiplatelet agents such as Revacept, represents a safe strategy to mitigate vascular restenosis. Revacept, a dimeric fusion protein of the extracellular domain of GPVI and the human Fc-fragment, inhibits collagen-mediated platelet adhesion and subsequent aggregation at the site of vascular injury. Finally, the possible utility of this pharmacological approach in the prevention of tumor metastasis is discussed.
Keywords
Revacept, GPVI, PTCA, Restenosis, Platelets, Collagen, TXA2, Aspirin, Tumor Metastasis
Introduction
Nearly 150 million individuals have coronary artery disease (CAD) worldwide, according to the latest Global Burden of Disease (GBD) dataset, with the highest prevalence in central and eastern Europe [1] and with a continuous rise in terms of absolute numbers [1].
Percutaneous transluminal coronary angioplasty (PTCA) also called percutaneous coronary intervention (PCI) represents the most common, minimally invasive procedure for resolving the narrowing or occlusion of the affected coronary artery, thus restoring an adequate blood supply to the injured tissues. Initially, PCI was performed using balloon catheters alone. However, due to subclinical outcomes and vessel restenosis, other devices were introduced including atherectomy devices and coronary stents. Coronary stents are the most widely used intracoronary devices in PTCA due to improved clinical outcomes. The procedure is mainly carried out by inserting stents into the damaged artery using specific fluoroscopic and/or endoscopic guidance [2]. Moreover, stents cause a decrease in vessel remodeling and elastic recoil at the surgery site [3-5]. Stent design, material, and strut thickness are objects of constant and continuous research and innovation. However, despite introducing new generations of stents to overcome drawbacks linked to both the medical procedure and the vascular biocompatibility [6], many patients continue to experience restenosis following the implantation.
Restenosis constitutes an excessive coronary artery response to damage during angioplasty and is characterized by early platelet activation, recruitment of inflammatory cells, the proliferation of vascular smooth muscle cells (VSMCs), and migration into the intima. VSMCs change their phenotype from contractile to synthetic, and this promotes extracellular matrix synthesis [7]. All these events characterize the phenomenon of neointimal hyperplasia.
To counteract the risk of either recurrent events or stent thrombosis, patients undergoing PCI are recommended to receive dual antiplatelet therapy (DAPT) [8] based on the combined use of low-dose Aspirin, which acts via selective inhibition of platelet cyclooxygenase (COX)-1 activity [9,10] and P2Y12 receptor antagonists [11]. Clopidogrel is the recommended P2Y12 antagonist in stable CAD, whereas prasugrel and ticagrelor represent the choice (if no contraindications are present) in patients with acute artery coronary syndrome [12]. However, this therapeutic approach is still associated with periprocedural ischemic events, representing an unfavorable prognostic factor and risk of bleeding [13]. Intense ongoing research is carried out to develop new antiplatelet agents affecting platelet aggregation and adhesion to damaged vessels without substantially altering hemostasis, thus reducing bleeding side-effects.
Revacept is a dimeric fusion protein of the extracellular domain of glycoprotein (GP) VI (the primary platelet collagen receptor) and the human Fc-fragment, which inhibits collagen-mediated platelet adhesion and subsequent aggregation at the site of vascular injury [14]. In this review, we have reported the pharmacological profile and clinical efficacy of Revacept [14,15] in cardiovascular medicine. Finally, the possible use of Revacept as an anticancer agent is discussed.
Structure and Signaling Pathways of the Platelet Surface Receptor Glycoprotein GPVI
At sites of vascular injury, extracellular matrix proteins, including collagen, are exposed and play a pivotal role in platelet activation and thrombosis [16]. Platelets express two primary receptors that directly bind collagen: integrin α2β1 (also known as VLA-2, GPIa-IIa, CD49) and GPVI [16]. GPVI was described as a platelet functional receptor by Moroi et al. in 1989 [17], who reported that a patient with mild bleeding time prolongation showed no platelet aggregation and adhesion induced by collagen but conserved a normal aggregation mediated by other agonists. Using SDS-PAGE/autoradiography technique, the authors showed that a 61-kD membrane glycoprotein (GP), identified as GPVI, was dramatically reduced, whereas GPs Ia, Ib, Ila, IIb, IIIa, and IV were normally expressed [17]. Interestingly, in the last years, some subjects with inherited or acquired defects of GPVI receptors have been described [18] and were characterized by a dramatic reduction of platelet response to collagen associated with only a slight prolongation of bleeding time and a normal platelet count [19,20].
GPVI is a single-chain protein of 60-65 kDa; it contains an extracellular chain with two collagen binding Ig-C2-like domains formed by disulfide bonds, a transmembrane region, and a 51 amino acid cytoplasmatic tail [21]. In its transmembrane region, GPVI harbors a positively charged arginine that permits a non-covalent association with the Fc receptor γ-chain (FcRγ) and its immunoreceptor tyrosine-based activation motif (ITAM). The cytosolic tail of GPVI contains a proline-rich motif that binds selectively to the SH3 domain of the Src family tyrosine kinases (SFK) that phosphorylates ITAM [22]. ITAM phosphorylation, in turn, triggers the recruitment and activation of the cytosolic tyrosine kinase Syk which allows the formation of the signaling complex [23] (Figure 1). This complex ultimately leads to the activation of phospholipase C (PLC)γ2, the major effector enzyme, causing the increase of intracellular calcium levels [Ca2+]. Following GPVI-mediated [Ca2+] increase, the small GTPase Rap1 is activated by CalDAG-GEFI and causes ERK/MAP kinase-dependent generation of thromboxane (TX)A2 [24]. TXA2, the primary product of arachidonic acid metabolism in activated platelets, promotes VSMC proliferation and migration [25-27]. GPVI is solely present in platelets and megakaryocytes [28]. In platelets, the interaction of GPVI to collagen leads to adhesion and aggregation by shifting integrins affinity state [including α2β1 (also known as VLA-2, GPIa-IIa, CD49b) and αIIbβ3 (also known as GPIIb -IIIa) (Figure 1).
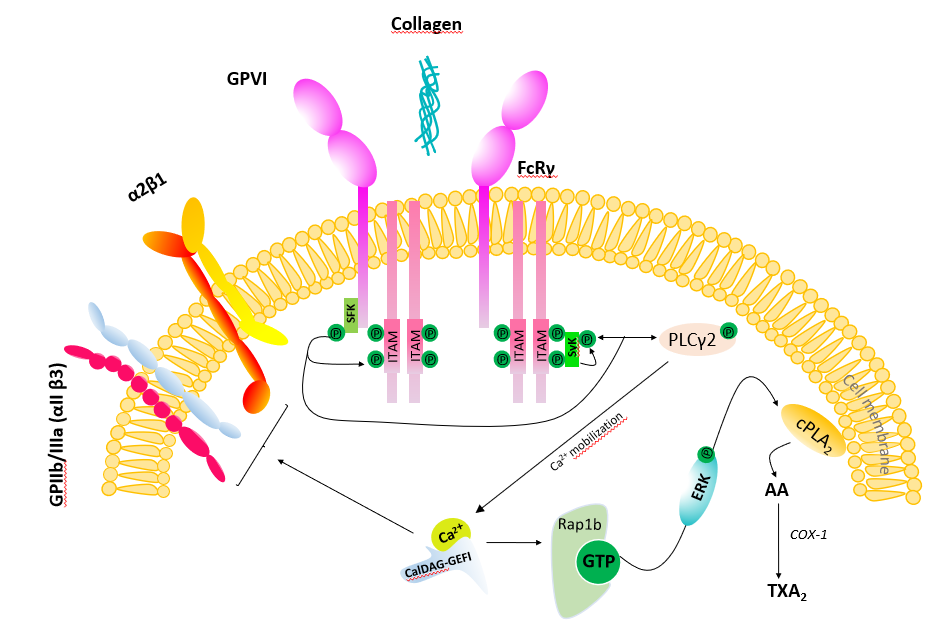
GPVI activation promotes the initial signaling cascade that leads to thrombus formation at sites of arterial injury or plaque rupture [16,29]. Morphologically diverse collagen type I- and collagen type III-containing structures in lipid-rich atherosclerotic plaques stimulate thrombus formation by activating platelet GPVI [30]. Human atheromatous plaque components involved in the development of atherothrombosis are collagen and tissue factor (TF). However, the first and rapid event in atherothrombosis is platelet adhesion and aggregation to plaque collagen via GPVI; the specific targeting of this first step is crucial and sufficient to inhibit atherothrombosis formation [31].
Revacept Pharmacological Profile
Revacept, a dimeric fusion protein of the extracellular domain of GPVI, is a novel, lesion-directed antithrombotic drug that does not interfere with the function of circulating platelets [14]. A phase I clinical study was performed to assess the safety, pharmacokinetics, and pharmacodynamics profile of Revacept [14]. In this study, 30 white subjects divided into five groups received 10, 20, 40, 80, and 160 mg of the drug in a single intravenous administration. The results demonstrated that the drug is safe [14]. Although Revacept serum concentrations decreased rapidly, the elimination rate was slow, with 1 mg/ml detected after two weeks in the serum of those subjects who had received the dose of 160 mg [14] (Table 1). The volume of distribution indicated that Revacept is confined to the systemic circulation (Table 1). Revacept dose-dependently inhibited collagen-induced platelet activation. The effect was detected 2 hrs after dosing with a maximum at 24 hrs. and lasted up to 7 days at the doses of 40-160 mg [14]. Platelet aggregation returned to basal conditions after two to three weeks depending on the dose of Revacept. Revacept did not affect ADP or thrombin-induced platelet aggregation.
| Revacept dose | |||||
|---|---|---|---|---|---|
| 10 mg | 20 mg | 40 mg | 80 mg | 160 mg | |
| AUC(0-t) (μg×h/mL) | 80.8 ± 23.3 | 187.9 ± 101.0 | 515.8 ± 120.6 | 898.0 ± 184.6 | 2330.3 ± 245.3 |
| Cmax (μg/mL) | 2.9 ± 1.1 | 5.1 ± 1.2 | 11.4 ± 2.4 | 20.0 ± 1.8 | 44.1 ± 3.6 |
| tmax (hrs) | 2.1 ± 3.8 | 1.3 ± 1.8 | 0.9 ± 0.8 | 0.9 ± 0.8 | 0.6 ± 0.2 |
| t1/2 (hrs) | 67.7 ± 6.5 | 87.5 ± 22.7 | 129.7 ± 7.9 | 137.6 ± 27.2 | 136.6 ± 36.7 |
| kel (1/hrs) | 0.010 ± 0.001 | 0.008 ± 0.002 | 0.005 ± 0.000 | 0.005 ± 0.001 | 0.005 ± 0.001 |
| CL (mL/min) | 1.8 ± 0.5 | 1.8 ± 0.64 | 1.3 ± 0.33 | 1.5 ± 0.3 | 1.1 ± 0.1 |
| Revacept was infused intravenously over 20 minutes. Values are reported as mean ± SD for 6 volunteers per dose group; AUC = the area under the plasma curve from time 0 to the last experimental time point [AUC(0-t)]; Cmax = the maximum plasma drug concentration; Tmax = the time to maximum plasma concentration, t1/2 (hrs) = terminal half-life of the drug in serum, Kel and CL represent elimination rate constant and total clearance of drug respectively | |||||
Other therapeutic strategies are under investigation to target GPVI protein. These are mainly based on the use of specific antibodies. Among these, Glenzocimab (ACT017), a humanized monoclonal antigen-binding antibody fragment F(ab) 9O12 that blocks GPVI function, is currently under evaluation in a phase II clinical study for the acute treatment of ischemic stroke in addition to the best emergency standard of care (including fibrinolysis by rtPA with or without added thrombectomy [32; Acute Ischemic Stroke Interventional Study (ACTIMIS) trial - NCT03803007)].
Compared to anti-GPVI-specific antibodies, Revacept shows the advantages of the local plaque-specific platelet inhibition without general GPVI blockage of circulating platelet with the potential risk of inducing platelet activation or GPVI deficiency, nor causing immunogenicity [14].
In vivo Studies Targeting Platelet GPVI Receptor
Previous studies in mice reported that the dimeric GPVI-Fc fusion protein inhibits platelet adhesion and aggregation [33] (Table 2). Moreover, Schonberger and colleagues demonstrated that the administration of GPVI-Fc fusion protein reduced infarct size, saving the cardiac function in a model of myocardial infarction induced by transient ligation of the left anterior descending artery [34] (Table 2). Repeated doses of Revacept led to a significant improvement of endothelial dysfunction and vascular morphology in an animal model of atherosclerosis (in rabbits); Revacept did not influence bleeding time alone or in combinations with various antiplatelet drugs [35] (Table 2). In addition, Revacept, in combination with low dose rtPA (0.35 mg/kg), was effective in improving reperfusion, reducing cerebral damage, and ameliorating neurological outcome, compared to rtPA given alone after experimental arterial thrombosis in a stroke mice model [36] (Table 2). Also, in this case, no increased bleeding was reported [36]. Revacept, added in vitro on top of Aspirin and the P2Y12 antagonist ticagrelor, increased the inhibition of platelet activation by atherosclerotic plaque [37,38]. GPVI-Fc alone or combined with Aspirin or ticagrelor did not increase closure time measured by the platelet function analyzer (PFA)-200. In detail, GPVI-Fc added on top of abciximab, a clinically used anti-fibrinogen receptor (GPIIb-IIIa) antibody which blocks platelet aggregation, also strongly inhibited stable (89%) platelet adhesion suggesting that not only transient adhesion is inhibited by Revacept [37].
| Animal model | Dose | Effect | References |
|---|---|---|---|
| C57BL/6 mice with carotid artery endothelial denudation | 1 mg/kg or 2 mg/kg GPVI-Fc or Fc control | Inhibition of platelet adhesion and arterial thrombus formation; Moderate prolongation of tail bleeding |
[31] |
| C57BL/6 mice with transient ligation of the left anterior descending artery (LAD) | 10 µg/g body or Fc control | Reduction of infarct size and preserved cardiac function | [32] |
| Rabbits with carotid artery endothelial denudation | 0.2 mg/kg; 0.6 mg/kg; 1 mg/kg; 2 mg/kg; and 3 mg/kg of Revacept or 2 mg/kg Fc control | Reduction of thrombus formation and a significant improvement of endothelial dysfunction and vessel wall thickness without increase bleeding | [33] |
| C57Bl/6J mice with ischemic stroke | 1 mg/kg Revacept in combination with 0.1 or 0.35 mg/ kg rtPA (Actilyse) or Fc control | Improved reperfusion, grip strength, infarct- surrounding edema without increasing intracranial bleeding | [34] |
Recently, we performed an in vivo study using Revacept [39] to test whether it could attenuate neointimal formation in a mouse model and prevent increased systemic TXA2 biosynthesis, which occurs after arterial injury [40,41]. In the early phase of restenosis, platelets adhere to VSMCs [42,43], at the site of vascular damage, due to subendothelial collagen exposure and release several soluble factors, including TXA2 [40,41,44]. This prostanoid is a potent stimulus for platelet aggregation, inducing vasoconstriction, endothelial adhesion molecule expression, and VSMC migration and proliferation [45]. In patients undergoing PCI, increased systemic TXA2 biosynthesis (assessed by measuring urinary levels of a major enzymatic metabolite, TXM) was largely inhibited by the use of low-dose Aspirin [40,41], i.e., a selective inhibitor of platelet COX-1-dependent TXA2 biosynthesis [9]. These results suggest that enhanced systemic TXA2 biosynthesis was caused by platelet activation at the site of vascular injury [45]. Enhanced systemic TXA2 biosynthesis was also found in the femoral artery wire injury model in C57BL/6 mice [39]. Urinary TXM levels increased at three days after vascular injury, compared to the values measured before the surgical treatment, and returned to control values at 28 days after injury (remodeling phase). The administration of Revacept, but not that of recombinant human IgG1 Fc (used as control), at a dose of 2 mg/kg/day from 3 days before until 7 days after injury, prevented the lesion-induced increase in urinary TXM levels at three days after vascular injury. These data suggest that platelet activation occurred in response to the endothelial damage due to the exposure of extracellular matrix proteins, such as collagen. Performing immunohistochemistry of femoral artery sections collected in the remodeling phase (at 28 days after injury), we found an increase of Ki-67 and CD68, which are recognized protein markers of cell proliferation and macrophage infiltration, respectively [39]. Revacept prevented these changes, and this was associated with the reduction of enhanced intima-to-media ratio in response to transluminal wire injury [39]. Our results show that Revacept, an inhibitor of the binding of platelet collagen receptors (mainly GPVI) to collagen exposed in areas of damaged endothelium [14] constrains the early release of TXA2 from activated platelets, promoting numerous cellular events that contribute to neointimal hyperplasia. Vascular remodeling and neointima formation was also strongly inhibited after endothelial damage in atherosclerotic ApoE -/- mice by Revacept confirming the robust effect on restenosis mechanisms induced by smooth muscle cells [46].
Effects of Antiplatelet Agents on the Crosstalk of Platelets with VSMCs
COX-2, an inducible enzyme that mediates the generation of prostanoids in inflammation [25], plays a role in restenosis progression through the generation of prostaglandin (PG)E2 and the activation of the PGE2 receptor subtype EP3α/β, and its signaling pathways cAMP/protein kinase A and phosphatidylinositol 3-kinase [47].
Alberti et al. [39] performed cocultures of human platelets and coronary artery smooth muscle cells (CASMCs) to address the hypothesis that platelet-derived TXA2 is the trigger of the induction of COX-2-derived-PGE2 in vascular restenosis. They found that platelet interaction with CASMCs caused an increase in TXB2 generation (the nonenzymatic product of TXA2) [39]. This was associated with an increased expression of COX-2 in CASMCs and enhanced release of PGE2 in the conditioned medium [39]. Rofecoxib, a selective COX-2 inhibitor [48], prevented the increase in PGE2 production detected in the cocultures suggesting a COX-2-dependent pathway activated by platelets in VSMCs. The role of platelet-derived TXA2 on COX-2 induction in CASMCs was assessed by pre-exposing platelets to Aspirin which was, then washed away before incubating platelets with CASMCs. Aspirin is an irreversible inhibitor of COX-isozymes [48], and since platelets do not have the nucleus and have limited de novo protein synthesis, the COX-1 inhibitory effect persists when the drug is removed. Under these experimental conditions, coculture TXB2 generation was profoundly inhibited [39]. In contrast, Rofecoxib did not affect it. Since platelets do not express COX-2, these data show that TXB2 was generated by platelet COX-1. Interestingly, it was found that PGE2 generated in the coculture was significantly reduced when aspirin-treated platelets were incubated with CASMCs. This effect was concomitant with the reduction of COX-2 expression in CASMCs. These results suggest that a platelet product (released from activated platelets and sensitive to Aspirin) is involved in inducing COX-2-dependent PGE2 in CASMCs exposed to platelets. Using a selective antagonist of the TXA2 receptor (TP), i.e., SQ 29,548, it was demonstrated that TXA2 is the trigger of COX-2 induction in CASMCs exposed to platelets [39]. Interestingly, Alberti and colleagues [39] found that the incubation of platelet-CASMC cocultures with Revacept, at clinically relevant concentrations, prevented the induction of COX-2 expression. Overall, these results show that platelets are activated by the interaction with VSMCs activates; then, released platelet TXA2 plays a central role in inducing COX-2-dependent PGE2 in VSMCs, considered a key mechanism in promoting vascular neointimal hyperplasia in response to mechanical injury [47]. COX-2 induction can be prevented by antiplatelet agents, such as Aspirin which affects platelet COX-1-dependent TXA2 biosynthesis, or by Revacept which prevents the interaction of platelets with VSMCs [39].
Selective COX-2 inhibitors (coxibs) can directly counteract COX-2-dependent PGE2 generation, in this setting; however, coxibs' pharmacological inhibition of vascular COX-2 is not recommended in this setting for their cardiovascular hazard [48].
Alberti et al. [39] have also shown that the interaction of platelets with CASMCs induced morphological changes in CASMCs, which assumed an epithelioid cell morphology, yielding a cobblestone pattern associated with the downregulation of α-SMA (α-smooth muscle actin) vs. CASMCs cultured alone; these changes describe the induction of the synthetic phenotype of vascular smooth muscle cells by the crosstalk with platelets. The downregulation of α-SMA and upregulation of COX-2 by the interaction of platelets with CASMCs were associated with the enhanced migratory capacity of CASMCs which was mitigated by the selective inhibition of platelet-TXA2 biosynthesis by Aspirin [39].
Collectively these findings sustain the distinct roles of platelets beyond their fundamental participation in primary hemostasis and support the use of novel and safer antiplatelet agents, such as Revacept, as a promising therapeutic strategy to prevent restenosis.
Clinical Studies with Revacept
Results of the first double-blind phase 2 clinical trial with Revacept in Patients with chronic coronary syndromes undergoing PCI
The ISAR-PLASTER study (The Intracoronary Stenting and Antithrombotic Regimen: Lesion Platelet Adhesion as Selective Target of Endovenous Revacept in Patients with Chronic Coronary Syndromes Undergoing Percutaneous Coronary Intervention) was designed to assess the safety and efficacy of Revacept (80 and 160 mg) and has been recently published [49]. The study enrolled 334 patients (mean age 67.4 years, 75.7% men and 27% diabetic) with stable ischemic heart disease undergoing elective PCI administered on top of standard DAPT. Three groups were identified based on the type of treatment: 120 subjects treated with Revacept at a dosage of 160 mg, 121 with Revacept at a dosage of 80 mg, 93 subjects with placebo. Patients received the drug in the form of a single intravenous infusion started immediately after the decision to perform angioplasty and in addition to standard antithrombotic therapy (Aspirin, Clopidogrel, Heparin, or Bivalirudin). The study's primary efficacy endpoint was a composite of death and myocardial damage defined as an increase in high sensitivity (HS) troponin, at least 5 times above the normal limit within 48 hrs of randomization. The following secondary endpoints were evaluated: the troponin HS peak at 48 hrs., all-cause mortality, spontaneous and periprocedural myocardial infarction, stroke, stent thrombosis, and urgent coronary revascularization within 30 days of randomization. The safety endpoint was bleeding (type 2 or higher according to the safety the Bleeding Academic Research Consortium-BARC) at 30 days. The patients were also subjected to measurements of platelet aggregation induced by ADP and collagen. The results show no significant differences in the incidence of the primary endpoint between the three groups. As regards the safety endpoint, no differences were found in the occurrence of BARC type 2 or higher bleeding at 30 days despite a significant added platelet inhibition by Revacept on top of DAPT. Thus, the authors conclude that Revacept administered on top of standard antithrombotic therapy is not associated with a reduction in periprocedural myocardial damage nor an increase in bleeding risk compared with placebo bleeding. This trial has several limitations. The enrolled population is small and at low risk of ischemic events; thus, the study does not have the statistical power to consider "hard" clinical endpoints.
Further, the post-procedural increase of troponin HS represents a clinical surrogate of myocardial damage with a low prognostic and long questioned scientific value [50-52]. However, the study confirms that Revacept appears to be a particularly safe drug, not associated with an increased risk of bleeding.
Results of a phase 2 study with Revacept in patients with symptomatic carotid stenosis
Another Phase 2 study was performed in 158 patients with symptomatic carotid stenosis with recent ischemic stroke or transient ischemic attack (TIA) in 16 centers in Germany and the United Kingdom (NCT01645306). All patients were on standard antiplatelet therapy and were treated with surgical carotid endarterectomy, carotid stenting, or best medical therapy according to the treatment guidelines. Fifty-one patients received placebo, 54 and 56 patients received 40 mg and 120 mg Revacept, respectively, as a single infusion on top of the standard antiplatelet medication. The study had exploratory endpoints investigating diffusion-weighted imaging by nuclear magnetic resonance (DWI-MRI) in a consecutive manner before study drug and directly after the carotid intervention to detect minor strokes which are partially clinically inapparent. As efficacy endpoints death, any stroke or TIA, myocardial infarction or need for PCI was investigated 30 days and 1 year after study drug application. Safety endpoints were bleeding rates according to the ReLy criteria. The study protocol has been published [53].
Patients with symptomatic carotid stenosis incur an up to 20% risk for a recurrent ischemic stroke within the first 3 months after an initial rather harmless TIA or minor stroke [54,55]. Moreover, carotid artery interventions such as stenting are accompanied with an up to 9% risk of peri-procedural stroke [56]. Therefore, these patients are jeopardized by the underlying vascular disease and the iatrogenic interventional procedure and are considered high risk patients.
The results of the study are not published yet, but positive effects are expected with no increase in bleeding complications. Therefore, Revacept seems the first plaque-specific thrombosis inhibitor without general influence on platelet function and hemostasis.
Further studies are needed to investigate whether in patients with the acute coronary syndrome, the peculiar mechanism of action of this drug, occurring at the level of plaque rupture, can be associated with a benefit in terms of reduction of ischemic events.
GPVI blockers in tumor metastasis
Several lines of evidence support the role of platelets in the pathogenesis of several inflammatory-based diseases, including cancer [57]. In cocultures of human colon carcinoma HT29 cells and platelets, Dovizio and colleagues have reported that unstimulated platelets interact rapidly with tumor cells through the binding of platelet collagen receptors (in particular, GPVI) and tumor components such as tumor components galectin-3 [58]. Galectin-3 is highly expressed in HT29 cells [58] and is unique among galectins because it contains a collagen-like domain. This early event translates into platelet activation, as demonstrated by the enhanced generation of TXA2. Direct platelet-tumor cell interaction was associated with enhanced mRNA expression of COX-2 and epithelial-mesenchymal transition (EMT)-inducing transcription factors, such as ZEB1 and TWIST1, and the mesenchymal marker VIM (vimentin). Later, platelet released platelet-derived growth factor (PDGF) which was associated with COX-2 mRNA stabilization via NHE-PI3K/PKCd-dependent nucleocytoplasmic translocation of the mRNA-stabilizing protein HuR [58]. In HT29 cells, overexpressed COX-2 and enhanced generation of PGE2 emanated mitogenic and survival signaling pathways through the downregulation of p21WAF1/CIP1 and the upregulation of cyclin B1as well as EMT inducing transcription factors and mesenchymal markers, such as vimentin, in association with repression of epithelial markers, such as E-cadherin. Revacept prevented the direct interaction of platelet and cancer cells, thus mitigating the induction of COX-2 and mRNA changes of EMT markers in platelet-HT29 cell cocultures [58]. These findings suggest that blockers of collagen-binding sites, such as Revacept, may represent an innovative strategy in colon cancer chemotherapy. Recently, in an orthotopic breast cancer model, GPVI deficiency was shown to reduce the number of spontaneously formed lung metastases compared with the wild-type controls [59]. The interaction of platelet GPVI with tumor cell galectin-3 played a critical role in platelet-induced tumor cell extravasation and metastasis [59].
In syngeneic mouse models of prostate and breast cancer, GPVI inhibition was also reported to cause tumor growth inhibition without compromising the physiological hemostasis process [60]. Moreover, this antiplatelet treatment favored the delivery of paclitaxel and liposomal doxorubicin to the tumor site, thus improving the therapeutic efficacy of these chemotherapeutics [60].
Concluding Remarks
Large amounts of evidence have accumulated on the role played by platelets in the development of human diseases, such as cardiovascular disease and cancer. Even though effective antiplatelet agents are available for clinical use, including Aspirin and P2Y12 antagonists, a great interest is in the development of safer agents associated with reduced bleeding complications. Revacept as a competitive GPVI inhibitor targeting collagen and other GPVI ligands is emerging as a promising lesion-specific, effective, and “bleeding-free” antiplatelet agent. The use of blockers of platelet GPVI can represent novel therapeutic strategies not only in cardiovascular medicine but also in fighting tumor metastasis.
Conflict of Interest
All authors declare none conflict of interest except Götz Munch, who is CEO of AdvanceCOR GmbH.
Funding Statements
This review was funded by Associazione Italiana per la Ricerca sul Cancro (AIRC) [IG 2017-ID. 20365 Project; Principal Investigator PP], by Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR) [Fondi per la Ricerca Scientifica di Ateneo, (ex 60%)] to PP and by Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR) [Fondi per la Ricerca Scientifica di Ateneo, (ex 60%)] to PB.
Author Contributions
Conceptualization: PP, GM; Writing-original draft preparation, PP, GM, AC, PB; Writing-review and editing, SA, PB, AC
Funding acquisition: PP, PB; All authors have read and agreed to the published version of the manuscript.
References
2. Beshchasna N, Ho AYK, Saqib M, Kraśkiewicz H, Wasyluk Ł, Kuzmin O, et al. Surface evaluation of titanium oxynitride coatings used for developing layered cardiovascular stents. Mater Sci Eng C Mater Biol Appl. 2019 Jun; 99:405-416.
3. Topol EJ, Serruys PW. Frontiers in interventional cardiology. Circulation.1998 Oct 27;98(17):1802-20.
4. Serruys PW, Regar E, Carter AJ. Rapamycin eluting stent: the onset of a new era in interventional cardiology. Heart. 2002 Apr;87(4):305-7.
5. Van den Brand MJ, Rensing BJ, Morel MA, Foley DP, de Valk V, Breeman A, et al. The effect of completeness of revascularization on event-free survival at one year in the ARTS trial. J Am Coll Cardiol. 2002 Feb 20;39(4):559-64.
6. Torii S, Cheng Q, Mori H, Lipinski MJ, Acampado E, Perkins LEL, et al. Acute thrombogenicity of fluoropolymer-coated versus biodegradable and polymer-free stents. EuroIntervention. 2019 Mar 20;14(16):1685-1693.
7. Weintraub WS. The pathophysiology and burden of restenosis. The American journal of cardiology. 2007 Sep 3;100(5):S3-9.
8. Sharma R, Kumar P, Prashanth SP, Belagali Y. Dual Antiplatelet Therapy in Coronary Artery Disease. Cardiol Ther. 2020 Dec;9(2):349-361.
9. Dovizio M, Bruno A, Tacconelli S, Patrignani P. Mode of action of aspirin as a chemopreventive agent. Recent Results Cancer Res. 2013; 191:39-65.
10. Picot D, Loll PJ, Garavito RM. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature. 1994 Jan 20;367(6460):243-9.
11. Mauri L, Kereiakes DJ, Yeh RW, Driscoll-Shempp P, Cutlip DE, Steg PG, et al. DAPT Study Investigators. Twelve or 30 months of dual antiplatelet therapy after drug-eluting stents. N Engl J Med. 2014 Dec 4;371(23):2155-66.
12. Sibbing D, Aradi D, Alexopoulos D, Ten Berg J, Bhatt DL, Bonello L, et al. Updated Expert Consensus Statement on Platelet Function and Genetic Testing for Guiding P2Y12 Receptor Inhibitor Treatment in Percutaneous Coronary Intervention. JACC Cardiovasc Interv. 2019 Aug 26;12(16):1521-1537.
13. Buccheri S, Capodanno D, James S, Angiolillo DJ. Bleeding after antiplatelet therapy for the treatment of acute coronary syndromes: a review of the evidence and evolving paradigms. Expert Opin Drug Saf. 2019 Dec;18(12):1171-1189.
14. Ungerer M, Rosport K, Bültmann A, Piechatzek R, Uhland K, Schlieper P, et al. Novel antiplatelet drug revacept (Dimeric Glycoprotein VI-Fc) specifically and efficiently inhibited collagen-induced platelet aggregation without affecting general hemostasis in humans. Circulation. 2011 May 3;123(17):1891-9.
15. Jamasbi J, Megens RT, Bianchini M, Uhland K, Münch G, Ungerer M, et al. Cross-Linking GPVI-Fc by Anti-Fc Antibodies Potentiates Its Inhibition of Atherosclerotic Plaque- and Collagen-Induced Platelet Activation. JACC Basic Transl Sci. 2016 Apr 25;1(3):131-142.
16. Nieswandt B, Watson SP. Platelet-collagen interaction: is GPVI the central receptor?. Blood. 2003 Jul 15;102(2):449-61.
17. Moroi M, Jung SM, Okuma M, Shinmyozu K. A patient with platelets deficient in glycoprotein VI that lack both collagen-induced aggregation and adhesion. J Clin Invest. 1989 Nov;84(5):1440-5.
18. Arthur JF, Dunkley S, Andrews RK. Platelet glycoprotein VI-related clinical defects. Br J Haematol. 2007 Nov;139(3):363-72.
19. Nurden AT. Clinical significance of altered collagen-receptor functioning in platelets with emphasis on glycoprotein VI. Blood Rev. 2019 Nov; 38:100592.
20. Hermans C, Wittevrongel C, Thys C, Smethurst PA, Van Geet C, Freson K. A compound heterozygous mutation in glycoprotein VI in a patient with a bleeding disorder. J Thromb Haemost 2009;7 (08):1356–1363.
21. Borst O, Gawaz M. Glycoprotein VI - novel target in antiplatelet medication. Pharmacol Ther. 2021 Jan;217:107630.
22. Dunster JL, Mazet F, Fry MJ, Gibbins JM, Tindall MJ. Regulation of Early Steps of GPVI Signal Transduction by Phosphatases: A Systems Biology Approach. PLoS Comput Biol. 2015 Nov 19;11(11):e1004589.
23. Rayes J, Watson SP, Nieswandt B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J Clin Invest. 2019 Jan 2;129(1):12-23
24. Stefanini L, Roden RC, Bergmeier W. CalDAG-GEFI is at the nexus of calcium-dependent platelet activation. Blood. 2009 Sep 17;114(12):2506-14.
25. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000
26. Hanasaki K, Nakano T, Arita H. Receptor-mediated mitogenic effect of thromboxane A2 in vascular smooth muscle cells. Biochem Pharmacol. 1990; 40:2535–2542.
27. Yokota T, Shiraishi R, Aida T, Iwai K, Liu NM, Yokoyama U, et al. Thromboxane A(2) receptor stimulation promotes closure of the rat ductus arteriosus through enhancing neointima formation. PLoS One. 2014 Apr15;9(4):e94895.
28. Clemetson JM, Polgar J, Magnenat E, Wells TN, Clemetson KJ. The platelet collagen receptor glycoprotein VI is a member of the immunoglobulin superfamily closely related to FcalphaR and the natural killer receptors. J Biol Chem. 1999 Oct 8;274(41):29019-24.
29. Ruggeri ZM. Platelets in atherothrombosis. Nat Med. 2002 Nov;8(11):1227-34.
30. Penz S, Reininger AJ, Brandl R, Goyal P, Rabie T, Bernlochner I, et al. Human atheromatous plaques stimulate thrombus formation by activating platelet glycoprotein VI. The FASEB Journal. 2005 Jun;19(8):898-909.
31. Reininger AJ, Bernlochner I, Penz SM, Ravanat C, Smethurst P, Farndale RW, et al. A 2-step mechanism of arterial thrombus formation induced by human atherosclerotic plaques. J Am Coll Cardiol. 2010 Mar 16;55(11):1147-58.
32. Lebozec K, Jandrot-PerrusM, Avenard G, Favre-Bulle O, Billiald P. Design, development and characterization of ACT017, a humanized Fab that blocks platelet’s glycoprotein VI function without causing bleeding risks. MAbs 2017;9(06):945–958.
33. Massberg S, Konrad I, Bültmann A, Schulz C, Münch G, Peluso M, et al. Soluble glycoprotein VI dimer inhibits platelet adhesion and aggregation to the injured vessel wall in vivo. FASEB J. 2004 Feb;18(2):397-9.
34. Schönberger T, Ziegler M, Borst O, Konrad I, Nieswandt B, Massberg S, et al. The dimeric platelet collagen receptor GPVI-Fc reduces platelet adhesion to activated endothelium and preserves myocardial function after transient ischemia in mice. Am J Physiol Cell Physiol. 2012 Oct 1;303(7):C757-66.
35. Ungerer M, Li Z, Baumgartner C, Goebel S, Vogelmann J, Holthoff HP, et al. The GPVI-Fc fusion protein Revacept reduces thrombus formation and improves vascular dysfunction in atherosclerosis without any impact on bleeding times. PLoS One. 2013 Aug 12;8(8):e71193.
36. Reimann A, Li Z, Goebel S, Fassbender J, Holthoff HP, Gawaz M, et al. Combined administration of the GPVI-Fc fusion protein Revacept with low-dose thrombolysis in the treatment of stroke. Heart Int. 2016 Apr 25;11(1):e10-e16.
37. Jamasbi J, Mergens RT, Bianchini M, Münch G, Ungerer M, Faussner A, et al. Differential inhibition of human atherosclerotic plaque- induced platelet activation by dimeric GPVI-Fc or anti-GPVI anitbodies. J Am Coll Cardiol 2015, 65: 2404-14
38. Mojica AK, Jamasbi J, Uhland K, Degen H, Münch G, Ungerer M, et al. Recombinant GPVI-Fc added to single ordual antiplatelet therapy in vitro prevents plaque-induced platelet thrombus formation. Thromb Haemost. 2017 Aug 1;117(8):1651-1659.
39. Alberti S, Zhang Q, D'Agostino I, Bruno A, Tacconelli S, Contursi A, et al. The antiplatelet agent revacept prevents the increase of systemic thromboxane A2 biosynthesis and neointima hyperplasia. Sci Rep. 2020 Dec 8;10(1):21420.
40. Ciabattoni G, Ujang S, Sritara P, Andreotti F, Davies G, Simonetti BM, et al. Aspirin, but not heparin, suppresses the transient increase in thromboxane biosynthesis associated with cardiac catheterization or coronary angioplasty. J Am Coll Cardiol. 1993 May;21(6):1377-81.
41. Braden GA, Knapp HR, FitzGerald GA. Suppression of eicosanoid biosynthesis during coronary angioplasty by fish oil and Aspirin. Circulation. 1991;84:679–685
42. Denes L, Entz L, Jancsik V. Restenosis and therapy. Int J Vasc Med. 2012;2012:406236.
43. Chandrasekar B, Tanguay JF. Platelets and restenosis. Am Coll Cardiol. 2000;35:555–562.
44. Kearney D, Byrne A, Crean P, Cox D, Fitzgerald DJ. Optimal suppression of thromboxane A(2) formation by Aspirin during percutaneous transluminal coronary angioplasty: no additional effect of a selective cyclooxygenase-2 inhibitor. J Am Coll Cardiol. 2004;43:526–531
45. Smyth EM. Thromboxane and the thromboxane receptor in cardiovascular disease. Clin Lipidol. 2010 Apr 1; 5(2): 209–219.
46. Schönberger T, Siegel-Axel D, Bußl R, Richter S, Judenhofer MS, Haubner R, et al. The immunoadhesin glycoprotein VI-Fc regulates arterial remodelling after mechanical injury in ApoE-/- mice. Cardiovascular research. 2008 Oct 1;80(1):131-7.
47. Zhang J, Zou F, Tang J, Zhang Q, Gong Y, Wang Q, et al. Cyclooxygenase-2-derived prostaglandin E₂ promotes injury-induced vascular neointimal hyperplasia through the E-prostanoid 3 receptor. Circ Res. 2013 Jul 5;113(2):104-14.
48. Patrignani P, Patrono C. Cyclooxygenase inhibitors: From pharmacology to clinical read-outs. Biochim Biophys Acta. 2015 Apr;1851(4):422-32.
49. Mayer K, Hein-Rothweiler R, Schüpke S, Janisch M, Bernlochner I, Ndrepepa G, et al. Efficacy and Safety of Revacept, a Novel Lesion-Directed Competitive Antagonist to Platelet Glycoprotein VI, in Patients Undergoing Elective Percutaneous Coronary Intervention for Stable Ischemic Heart Disease: The Randomized, Double- blind, Placebo-Controlled ISAR-PLASTER Phase 2 Trial. JAMA Cardiol. 2021 Jul 1;6(7):753-761.
50. Sherwood MW, Kristin Newby L. High-sensitivity troponin assays: evidence, indications, and reasonable use. J Am Heart Assoc. 2014;3(1):e000403.
51. Garcia-Garcia HM, McFadden EP, von Birgelen C, Rademaker-Havinga T, Spitzer E, Kleiman NS, et al. Impact of Periprocedural Myocardial Biomarker Elevation onMortality Following Elective Percutaneous Coronary Intervention. JACC Cardiovasc Interv. 2019 Oct14;12(19):1954-1962.
52. Raber I, McCarthy CP, Januzzi JL Jr. A Test in Context: Interpretation of High-Sensitivity Cardiac Troponin Assays in Different Clinical Settings. J Am Coll Cardiol. 2021 Mar 16;77(10):1357-1367.
53. Gröschel K, Uphaus T, Loftus I, Poppert H, Diener HC, Zobel J, et al. Revacept, an inhibitor of platelet adhesion in symptomatic carotid artery stenosis: Design and rationale of a randomized phase 2 clinical trial. TH Open 2020; 4: e393-e399
54. Johnston SC, Gress DR, Browner WS, Sidney S. Short-term prognosis after emergency department diagnosis of TIA. JAMA. 2000 Dec 13;284(22):2901-6.
55. Lovett JK, Dennis MS, Sandercock PA, Bamford J, Warlow CP, Rothwell PM. Very early risk of stroke after a first transient ischemic attack. Stroke. 2003 Aug;34(8):e138-40.
56. Carotid Stenting Trialists' Collaboration, Bonati LH, Dobson J, Algra A, Branchereau A, Chatellier G, Fraedrich G, et al. Short-term outcome after stenting versus endarterectomy for symptomatic carotid stenosis: a preplanned meta-analysis of individual patient data. Lancet. 2010 Sep 25;376(9746):1062-73.
57. Dovizio M, Ballerini P, Fullone R, Tacconelli S, Contursi A, Patrignani P. Multifaceted Functions of Platelets in Cancer: From Tumorigenesis to Liquid Biopsy Tool and Drug Delivery System. Int J Mol Sci. 2020 Dec 16;21(24):9585.
58. Dovizio M, Maier TJ, Alberti S, Di Francesco L, Marcantoni E, Münch G, et al. Pharmacological Inhibition of platelet-tumor cell crosstalk prevents platelet-induced overexpression of cyclooxygenase-2 in HT29 human colon carcinoma cells. Mol Pharmacol. 2013 Jul;84(1):25-40.
59. Mammadova-Bach E, Gil-Pulido J, Sarukhanyan E, Burkard P, Shityakov S, Schonhart C, et al. Platelet glycoprotein VI promotes metastasis through interaction with cancer cell-derived galectin-3. Blood. 2020 Apr 2;135(14):1146-1160.
60. Volz J, Mammadova-Bach E, Gil-Pulido J, Nandigama R, Remer K, Sorokin L, et al. inhibition of platelet GPVI induces intratumor hemorrhage and increases efficacy of chemotherapy in mice. Blood. 2019 Jun 20;133(25):2696-2706.