Abstract
Results of epidemiological studies show that obesity and type 2 diabetes mellitus have become a public health concern globally, which has substantial health, social and economic impacts. A common characteristic of human obesity and type 2 diabetes is insulin resistance, which a given amount of insulin produces less than normal physiological responses, usually demonstrated as diminished glucose lowering effect of insulin. Daily feeding cycles lead to the variations of nutrient and hormone levels. Insulin from pancreatic β-cells acts on the liver to control short-term and long-term metabolic homeostasis. The long-term effects of insulin can be attributed, partially, to insulin-regulated expressions of genes involved in glucose and lipid metabolism. In an insulin-sensitive liver, insulin suppresses expressions of gluconeogenic genes, and increases expressions of lipogenic genes. In an insulin-resistant liver, insulin fails to suppress expressions of gluconeogenic genes, while expression levels of lipogenic genes are elevated. This may cause a vicious cycle that drives β-cell failure and over diabetes. Using sterol-regulatory element binding protein 1c (Srebp-1c) and phosphoenolpyruvate carboxykinase (Pck1) genes as model genes of insulin-regulated expression, we found that insulin failed to regulate their expressions in primary hepatocytes from Zucker fatty rats fed ad libitum, and Zucker lean rats after over-eating. For the first time, we observed the presence of hepatic insulin resistance at gene expression (HIRAGE). This review was aimed to summarize the current understanding of insulin-regulated hepatic gene expression. It focuses on the potential roles of nutrient fluxes in the development of HIRAGE, which is supported by clinical observations of bariatric surgery studies. It argues that a transient and dynamic HIRAGE exists after overnutrition and precedes a systemic insulin resistance in a wildtype animal. The underlying mechanisms of HIRAGE may help us to identify intervention points for the prevention and treatment of hepatic insulin resistance and type 2 diabetes.
Keywords
Insulin; Liver; Hepatic insulin resistance; Gene expression; Diabetes; Bariatric surgery
Introduction
The International Diabetes Federation reported 463 million diabetic adults globally in 2019 [1]. In the United States alone, 34.1 million people have diabetes according to the National Diabetes Statistical Report 2020 by the Center for Disease Control and Prevention [2]. These numbers can increase further in the future [3,4]. The high obesity prevalence in the United States population [5] also predicts a rise of patients with type 2 diabetes mellitus in the future [6], showing the risk in children and adolescents [7]. Due to morbidity and mortality of diabetes, the social and economic costs have been substantial [8,9]. With the development of biotechnology, obese gene [10], and other genes potentially contributing to type 2 diabetes [11] have been gradually determined. In addition, environmental factors such as gut microbiota have also been implied for chronic diseases [12]. Another important factor for type 2 diabetes is nutrition, the sum of all the biochemical reactions occurring to nourish an organism, showing the multifactorial nature of type 2 diabetes.
The history of understanding diabetes is a long list of sentimental discoveries [13,14]. The early studies linked the pancreas (an organ) to diabetes (a disease), and then insulin from the pancreas (a hormone) to the treatment of diabetes. These have shaped our views of development of not only diabetes, but also other diseases in general [14]. Two types of patients requiring dramatically different amounts of insulin to control glucose indicated two types of diabetes, sensitive and resistant ones [15]. This happened more than two decades before the radioimmunoassay of insulin [16]. Patients with type 2 diabetes are insulin resistant, which a given amount of insulin produces less than normal physiological effects [17].
Insulin resistance has been studied extensively at systemic, organ, tissue and cellular and molecular levels [18,19]. Overnutrition plays an essential role in the development of chronic metabolic diseases such as obesity and type 2 diabetes. For subjects without genetic defects, the development of insulin resistance and type 2 diabetes is a graduate process. How the transition from an insulinsensitive state to an insulin-resistant state occurs, and what the roles of nutrients are in the process have not been fully understood. Here, we try to summarize the current understanding of insulin-regulate gene expression in the liver, and describe a phenomenon of hepatic insulin resistance at gene expression (HIRAGE), which may be linked to overnutrition.
Insulin Signaling and Resistance
As shown in Figure 1, insulin secreted from pancreatic β-cells after feeding regulates glucose, lipid and protein metabolism [20]. It acts with dietary nutrients on the liver first via portal vein and then to other parts of the body [21]. As shown in Figure 2, insulin signal is mediated by its receptor in the cell membrane and intracellular components of signal transduction cascades [22]. The signaling transduction begins when insulin binds to its cell surface receptor [23], which is followed by phosphorylation and dephosphorylation of proteins in cytosol and membranes [22,24]. For example, insulin receptor tyrosine kinase phosphorylates insulin receptor substrates [25], which recruit additional proteins through protein-protein interactions to transduce insulin signal [26]. These proteins include the regulatory subunits of phosphatidylinositol 3-kinase, growth factor receptorbound protein 2, and protein tyrosine phosphatase-2 [27]. Additional players include member in phosphatidylinositol 3-kinase/protein kinase B (also known as Akt), and GRB2/ mitogen activated protein kinase pathways [28-30]. The coordinative actions of these players eventually lead to short-term (in minutes) and long-term (in hours) changes of activities and expression levels of proteins, respectively.
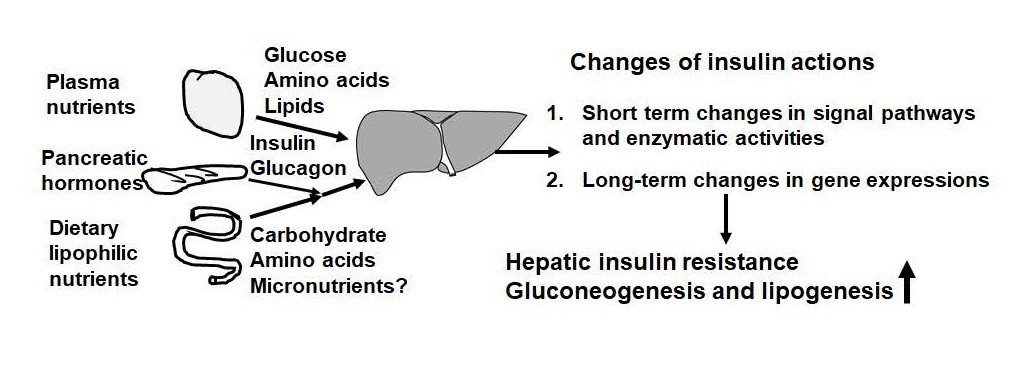
Figure 1. Nutritional and hormonal factors regulating liver metabolism and insulin action in it. Food intakes lead to rise and fall of plasma levels of nutrients (glucose, amino acids, and lipids) and pancreatic hormones (insulin and glucagon) every day. Insulin stimulation regulates the enzyme activities in the short-term through modifications and in the long-term via gene expressions. The development of obesity and type 2 diabetes is associated with insulin resistance in the liver, which shows elevations of hepatic gluconeogenesis and lipogenesis.
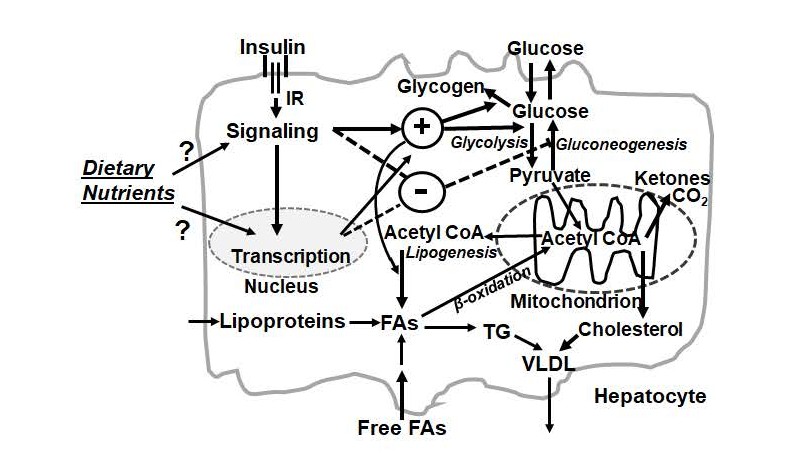
Figure 2. Potential impact points of dietary nutrients on insulin actions in a hepatocyte. The influxes of dietary nutrients trigger entries of nutrients into the liver and secretion of insulin. Insulin binds to its receptor (IR) to initiate a signaling pathway that can regulate glucose and lipid metabolism via alteration of enzymatic activities in the short-term and gene expression in the long-term. For glucose metabolism, insulin increases conversions of glucose into pyruvate in glycolysis and into glycogen. At the same time, the hepatic gluconeogenesis is suppressed. Pyruvate is converted into acetyl CoA, which is used for the synthesis fatty acids (FAs) and cholesterol. FAs can come from lipogenesis, free FAs and lipoproteins in the plasma. FAs can be utilized through β-oxidation to generate acetyl CoA, and then ketones. Insulin promotes lipogenesis that converts glucose into FAs. FAs are esterified into triacylglycerol (TG). TG and cholesteryl esters are incorporated into very low-density lipoprotein (VLDL) which is secreted from the hepatocytes and delivered to other parts of the body. Dietary nutrients may modulate insulin signal transduction and insulin-regulated transcription in a hepatocyte. These points deserve to be studied in the future.
The evaluation of insulin sensitivity and resistance has been relied on ability of insulin to control blood glucose. In vivo, insulin secretion and sensitivity/resistance have been calculated by clamp techniques that usually last 2 hours [31]. Glucose is infused to achieve hyperglycemia for the measurement of plasma insulin level. To determine insulin sensitivity, insulin is infused while glucose level is held at constant for the estimations of glucose usage in peripheral tissues and the hepatic glucose production [31]. The hyperinsulinemic-euglycemic clamp is used to estimate hepatic glucose production by calculating the rates of glucose uptake and metabolism in individual tissues [32]. Additionally, rapid insulin sensitivity test, and intravenous insulin tolerance test have been used to determine insulin sensitivity [33]. These methods allow the measurements of those parameters accurately within the 2-hour window. Another popular method using fasting glucose and insulin data is called homeostatic model assessment [34].
Recently, real-time and continuous blood glucose monitoring has been used for patients in non-hospital settings. The United States Food and Drug Administration approved the first continuous glucose monitoring system for diabetic subjects without further confirmation with results from other devices in 2016 [35]. Several types of real-time continuous glucose monitoring with different characteristics are available for clinical uses [36]. They are helpful to manage glucose [37], and to apply personalized nutrition strategies in patients with type 2 diabetes [38].
The Hepatic Responses to Insulin
As shown in Figure 1, the liver receives blood flows from the portal vein bringing dietary nutrients from the gastrointestinal tract and hormones from pancreas, and from the hepatic artery carrying chylomicron remnants and endogenous metabolites. Insulin works coordinatively with nutrients and other hormones to regulate the hepatic metabolism. For the short-term, insulin stimulation leads to covalent modifications and allosteric regulations of enzymes responsible for fuel metabolism [20,22].
For example, insulin alters the phosphorylation and dephosphorylation states of glycogen synthase and phosphorylase, which determine their activities to store and release glucose, respectively [39,40].
For the long-term, insulin regulates expressions of hepatic genes involved in glycolysis, glycogenesis, lipogenesis (lipid biosynthesis), and gluconeogenesis (glucose production) [41]. As shown in Figure 2, insulin works together with dietary signals to change the activation states of signaling transduction components and transcription of genes in the liver. The outcomes are the increases of glucose to be stored as glycogen and used to produce acetyl-CoA for the conversion into fatty acids and cholesterol, which are eventually incorporated into very-low density lipoprotein as triacylglycerol and cholesteryl esters, respectively. Insulin increases the expression of glucokinase gene (Gck) [42,43], the enzyme responsible for the first step of the hepatic glycolysis. It inhibits the expression of the cytosolic form of phosphoenolpyruvate carboxykinase gene (Pck1) [44] and glucose 6-phosphatase catalytic subunit gene (G6pc) [41], the first and last enzymes for gluconeogenesis, respectively. For the hepatic lipid metabolism, insulin increases the mRNA level of sterol regulatory elementbinding protein 1c gene (Srebp-1c) [45], a key inducer of hepatic lipogenesis [46].
The Hepatic Insulin Resistance at Gene Expression (HIRAGE)
The daily food intakes in meals bring macronutrients and micronutrients into the body. This causes rise and fall of plasma nutrient and hormone levels. The liver responds to these changes such as glucose and insulin accordingly, at least in part, through alterations of expression levels of hepatic genes involved in the glucose and lipid metabolism. These changes occur in a cyclic and balanced manner on a daily basis, indicating the transit and dynamic nature of the process. Depending on types and amounts of those nutrients, developmental stages of an organism and disease states, these changes may be disrupted either temporarily or permanently. When temporary changes are modified and allowed to last longer than the period of the daily feeding cycle, the nutrient influxes coming with the next feeding cycle may change the hepatic metabolism dramatically, and lead to systemic insulin resistance. When this occurs, interventions are needed to bring the cycle back or establish a new balance to prevent the progression of diseases.
With the development of insulin resistance, profound changes of hepatic lipid and glucose metabolism occur [17], which are associated with alterations of insulin-regulated hepatic genes [18,47,48]. In an insulin-sensitive liver, insulin suppresses genes for gluconeogenesis and increases those for glycolysis and lipogenesis (Figure 2). In an insulin-resistant liver, hyperinsulinemia fails to suppress expressions of gluconeogenic genes, but is associated with elevated expressions of lipogenic genes [17,18]. Here, the expression levels of both gluconeogenic and lipogenic genes are elevated [47]. This accelerates systemic insulin resistance as hyperglycemia and hyperlipidemia promotes more insulin secretion, which further induces lipogenesis and results in a vicious cycle, and in turn, leads to β-cell failure and over diabetes [17,18].
Nutritional, neuronal and hormonal factors can affect hepatic insulin actions directly and indirectly [49-53]. Insulin resistance can be caused by dietary factors such as fructose [17], supplementations of antioxidants [54], and fatty acids [55]. Pregnant female mice fed a high-fat diet generate offspring with hepatic insulin resistance [53]. Dietary glucose and amino acids, but not fat, stimulate a vagal reflex mediated by hepatic parasympathetic nerves to increase insulin sensitivity in rats [33]. Infusion of isoleucine or valine in hypothalamus lowers hepatic glucose production in rats, which is impaired by intake of a high-fat diet [56]. Resistin-induced neuropeptide Y in the lateral cerebral ventricle [57], and activation of p70 S6 kinase in hypothalamus of mice fed a high-fat diet [58] cause hepatic insulin resistance.
The liver specific insulin receptor knockout (LIRKO) mice have been used to investigate the role of insulin signaling in the liver [59,60]. LIRKO mice at age of 2 and 6 months have lower plasma TG and free fatty acid levels, and higher glucose level than the control mice [59]. Interestingly, during fasting, they have higher and lower blood glucose levels than the control ones at 2 and 6 months of age, respectively [59]. Only, the 2-month, but not 6-month, old LIKRO mice develop hepatic insulin resistance with elevated Pck1 and G6pc, and reduced Gck and liver type pyruvate kinase expression levels [59]. Interestingly, LIRKO and control mice have the same rate of hepatic glucose production [59]. The 8-10-week-old LIRKO mice have higher Srebp-1c mRNA expression than the control ones, and refeeding still partially induces Srebp-1c [61]. This suggests that nutrient fluxes contribute to the hepatic induction of Srebp-1c in LIRKO mice.
Insulin is needed for the hepatic lipogenesis after refeeding [62]. This was attributed to the insulin-induced expression Srebp-1c [45], a key regulator of hepatic lipogenesis [63]. Using primary rat hepatocytes, we have identified the insulin responsive element on the hepatic Srebp-1c promoter, which contains two liver X receptor elements and one sterol-regulatory element [64]. The two liver X receptor elements are also retinoic acid responsive elements [65], demonstrating the converge sites of nutritional and hormonal signals on Srebp-1c promoter.
As noted above, insulin has been responsible for metabolism of glucose and lipid. In insulin sensitive liver, the hepatic gene expressions involved in those processes are active and sensitive as well. However, upon insulin resistance, the genes mentioned are resistant accordingly in which we call hepatic insulin resistance at gene expression (HIRAGE). In our previous study, we have compared the insulin-regulated Pck1 and srebp-1c in primary hepatocytes from insulin-sensitive Zucker lean and insulin-resistant Zucker fatty rats [66]. Zucker fatty rats are obese and insulin resistant due to hyperphagia caused by mutations of leptin receptors [67-70]. Insulin failed to regulate Srebp-1c and Pck1 in hepatocytes from Zucker fatty, but not lean, rats fed ad libitum [66]. The excessive nutrient fluxes combined with hyperinsulinemia in Zucker fatty rats fed ad libitum cause HIRAGE. Interestingly, HIRAGE is partially attenuated in hepatocytes from Zucker fatty rats fasted for overnight [66]. These results demonstrate that HIRAGE is a dynamic and transit state associated with hyperphagia.
The Role of Nutrient Fluxes in HIRAGE in Insulin Sensitive Animals
Dietary factors can affect insulin action in the body, which initiated the concept of insulin resistance [71]. Reduction of energy and nutrient intakes leads to corrections of obesity and type 2 diabetes. A systematic review and meta-analysis of 11 randomized clinical trials shows that bariatric surgery procedures lead to improvements of parameters of obesity and type 2 diabetes in human subjects [72]. Recently, results of randomized clinical trials containing patients with type 2 diabetes have consistently shown superior efficacy of bariatric surgery in reducing weight and lowering blood glucose, compared to other medical and lifestyle interventions [73-80]. Therefore, surgery treatments have been recommended for type 2 diabetes patients with class III obesity and those with class II obesity and inadequately controlled blood glucose by other methods [81]. Addition to reductions of nutrient intakes, changes of gut hormones and gut-brain axis are also attributed to the benefits of bariatric surgery procedures [82].
We have conducted a pair-feeding study to evaluate the effects of energy and nutrient intakes on metabolism in Zucker lean and fatty rats fed a vitamin A deficient or sufficient diet for 8 weeks [83]. In the last 24-hour of the study, rats pairfed the isocaloric vitamin A sufficient diet were divided into two groups. Rats in first group were fed exactly the same amount of energy as those in the vitamin A deficient group, whereas vitamin A sufficient rats in the second group were fed ad libitum, which led to ~40% more intake of the diet than the first group. For the first time, HIRAGE was observed in primary hepatocytes from Zucker lean rats in the second, but not the first, pairfeeding group [83]. HIRAGE occurs in Zucker lean rats, who have wild type genome and are considered insulin-sensitive.
The bariatric surgery procedures can successfully reduce insulin resistance and manage glucose homeostasis in type 2 diabetes patients. The nutrient fluxes from the gastrointestinal tract to the liver (question marks in Figures 1 and 2) clearly cause HIRAGE. Whether the total energy, micronutrient(s), macronutrient(s) or their combinations or whether nutrient and hormone interactions contribute to HIRAGE remain to be determined.
Conclusions and Future Perspectives
Currently, the insulin-mediated suppression of hepatic glucose production and phosphorylation of signal cascade component such as phosphor-Akt Ser473 are great indicators of insulin actions to evaluate hepatic insulin resistance [20]. It is worth to note that players of insulin signaling pathway are not only for insulin. For example, phosphatidylinositol 3-kinase and mitogen activated protein kinase are also activated by platelet-derived growth factor [84]. Akt phosphorylation is a converge points of multiple receptor tyrosine kinases [85]. We have shown that the hepatic levels of phosphor-Akt Ser473 and The450 in Zucker fatty rats are higher than that in Zucker lean rats [86]. Insulin induces phosphor-Akt Ser473 and Thr308 similarly in primary hepatocytes from Zucker lean and fatty rats [86]. Given the dramatic changes of metabolism in the liver, phosphor-Akt Ser473 probably cannot serve as an indicator of HIRAGE. Clearly, more studies of insulinregulated gene expression are needed.
How dynamic changes of nutrient fluxes and insulin levels in a daily feeding cycle interact and modulate each other’s functions in the liver is still unclear. Nevertheless, HIRAGE occurs transiently and dynamically in a healthy animal only due to excessive nutrient fluxes. Does this mean that the liver has an insulin resistant state in a regular feeding cycle? Whether the HIRAGE is analogous to the hepatic insulin resistance in human patients or mechanically the same remains to be revealed. The dynamic and transient HIRAGE may occur long before the systemic insulin resistance happens, a topic that deserves to be explored. The underlying mechanisms of HIRAGE may provide multiple interventional targets for prevention and treatment of hepatic insulin resistance and type 2 diabetes. With more and more data from the use of continuous glucose monitoring [38], understanding HIRAGE may contribute to the practice of personalized nutrition to benefit patients with type 2 diabetes as well.
Acknowledgement
The authors would like to thank the Scientific Research Project of Wuhan Municipal Health Commission for research support to Y. Z. (Number: WX19Y09).
Author Contributions
Zhang Y and Chen G designed the outline and wrote the draft.
Conflict of Interest Statement
All authors declared no conflict of interest.
References
2. Centers for Disease Control and Prevention. National diabetes statistics report, 2020. Atlanta, GA: Centers for Disease Control and Prevention, US Department of Health and Human Services. 2020.
3. Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Research and Clinical Practice. 2014 Feb 1;103(2):137- 49.
4. Ogurtsova K, da Rocha Fernandes JD, Huang Y, Linnenkamp U, Guariguata L, Cho NH, et al. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Research and Clinical Practice. 2017 Jun 1;128:40-50.
5. Yanovski SZ, Yanovski JA. Obesity prevalence in the United States—up, down, or sideways?. New England Journal of Medicine. 2011 Mar 17;364(11):987-9.
6. Schulze MB, Hu FB. Primary prevention of diabetes: what can be done and how much can be prevented? Annu Rev Public Health. 2005 Apr 21;26:445-67.
7. Grossman DC, Bibbins-Domingo K, Curry SJ, Barry MJ, Davidson KW, Doubeni CA, et al. Screening for obesity in children and adolescents: US Preventive Services Task Force recommendation statement. JAMA. 2017 Jun 20;317(23):2417-26.
8. Care D. Economic Costs of Diabetes in the US in 2017. Diabetes care. 2018 May;41:917.
9. Cho N, Shaw JE, Karuranga S, Huang Y, da Rocha Fernandes JD, Ohlrogge AW, et al. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Research and Clinical Practice. 2018 Apr 1;138:271-81.
10. Friedman JM. Leptin at 14 y of age: an ongoing story. The American Journal of Clinical Nutrition. 2009 Mar 1;89(3):973S-9S.
11. Gaulton KJ, Willer CJ, Li Y, Scott LJ, Conneely KN, Jackson AU, et al. Comprehensive association study of type 2 diabetes and related quantitative traits with 222 candidate genes. Diabetes. 2008 Nov 1;57(11):3136-44.
12. Vallianou N, Stratigou T, Christodoulatos GS, Dalamaga M. Understanding the role of the gut microbiome and microbial metabolites in obesity and obesity-associated metabolic disorders: Current evidence and perspectives. Current Obesity Reports. 2019 Sep 15;8(3):317-32.
13. Vecchio I, Tornali C, Bragazzi NL, Martini M. The discovery of insulin: an important milestone in the history of medicine. Frontiers in Endocrinology. 2018 Oct 23;9:613.
14. Bliss M. The Discovery of Insulin: McClelland and Stewart Limited; 1982 1982.
15. Himsworth HP. Diabetes Mellitus. The Lancet. 1936;227(5864):2.
16. Yalow RS, Berson SA. Immunoassay of endogenous plasma insulin in man. The Journal of Clinical Investigation. 1960 Jul 1;39(7):1157-75.
17. McGarry JD. Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes. 2002 Jan 1;51(1):7-18.
18. Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metabolism. 2008 Feb 6;7(2):95-6.
19. McGarry JD. What if Minkowski had been ageusic? An alternative angle on diabetes. Science. 1992 Oct 30;258(5083):766-70.
20. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiological Reviews. 2018 Oct 1;98(4):2133-223.
21. Cherrington AD, Edgerton D, Sindelar DK. The direct and indirect effects of insulin on hepatic glucose production in vivo. Diabetologia. 1998 Aug 1;41(9):987- 96.
22. Cohen P. The twentieth century struggle to decipher insulin signalling. Nature Reviews Molecular Cell Biology. 2006 Nov;7(11):867-73.
23. McKern NM, Lawrence MC, Streltsov VA, Lou MZ, Adams TE, Lovrecz GO, et al. Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature. 2006 Sep;443(7108):218-21.
24. Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nature Reviews Molecular Cell Biology. 2006 Feb;7(2):85-96.
25. Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nature Reviews Molecular Cell Biology. 2006 Feb;7(2):85-96.
26. Hanke S, Mann M. The phosphotyrosine interactome of the insulin receptor family and its substrates IRS-1 and IRS-2. Molecular & Cellular Proteomics. 2009 Mar 1;8(3):519-34.
27. Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia. 2012 Oct 1;55(10):2565-82.
28. Avruch J. MAP kinase pathways: the first twenty years. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 2007 Aug 1;1773(8):1150-60.
29. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007 Jun 29;129(7):1261- 74.
30. Osborne JK, Zaganjor E, Cobb MH. Signal control through Raf: in sickness and in health. Cell Research. 2012 Jan;22(1):14-22.
31. DeFronzo RA, Tobin JD, Andres R. The glucose clamp technique: a method for the quantification of beta cell sensitivity to glucose and of tissue sensitivity to insulin. Am J Physiol. 1979;237(E214-E223):1.
32. Kim JK, Kim YJ, Fillmore JJ, Chen Y, Moore I, Lee J, et al. Prevention of fat-induced insulin resistance by salicylate. The Journal of Clinical Investigation. 2001 Aug 1;108(3):437-46.
33. Afonso RA, Gaspar JM, Lamarão I, Lautt WW, Macedo MP. Postprandial insulin action relies on meal composition and hepatic parasympathetics: dependency on glucose and amino acids: Meal, parasympathetics & insulin action. The Journal of Nutritional Biochemistry. 2016 Jan 1;27:70-8.
34. Tm W, Levy JC, Matthews DR. Use and abuse of HOMA modeling. Diabetes Care. 2004;27:1487-95.
35. Shapiro AR. FDA approval of nonadjunctive use of continuous glucose monitors for insulin dosing: a potentially risky decision. JAMA. 2017 Oct 24;318(16):1541-2.
36. Mian Z, Hermayer KL, Jenkins A. Continuous glucose monitoring: review of an innovation in diabetes management. The American Journal of the Medical Sciences. 2019 Nov 1;358(5):332-9.
37. Lu M, Zuo Y, Guo J, Wen X, Kang Y. Continuous glucose monitoring system can improve the quality of glucose control and glucose variability compared with point-of-care measurement in critically ill patients: A randomized controlled trial. Medicine. 2018 Sep;97(36).
38. Zeevi D, Korem T, Zmora N, Israeli D, Rothschild D, Weinberger A, et al. Personalized nutrition by prediction of glycemic responses. Cell. 2015 Nov 19;163(5):1079-94.
39. Agius L. Role of glycogen phosphorylase in liver glycogen metabolism. Molecular Aspects of Medicine.2015 Dec 1;46:34-45.
40. Hojlund K. Metabolism and insulin signaling in common metabolic disorders and inherited insulin resistance. Danish Medical Journal. 2014;61(7):B4890.
41. O’Brien R, Granner D. Regulation of gene expression by insulin. Physiological Reviews. 1996;76(4):1109-61.
42. Iynedjian PB. Mammalian glucokinase and its gene. Biochemical Journal. 1993 Jul 1;293(1):1-3.
43. Magnuson MA, Andreone TL, Printz RL, Koch S, Granner DK. Rat glucokinase gene: structure and regulation by insulin. Proceedings of the National Academy of Sciences. 1989 Jul 1;86(13):4838-42.
44. Hanson RW, Reshef L. Regulation of phosphoenolpyruvate carboxykinase (GTP) gene expression. Annual Review of Biochemistry. 1997 Jul;66(1):581-611.
45. Shimomura I, Bashmakov Y, Ikemoto S, Horton JD, Brown MS, Goldstein JL. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocininduced diabetes. Proceedings of the National Academy of Sciences. 1999 Nov 23;96(24):13656-61.
46. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. The Journal of Clinical Investigation. 2002 May 1;109(9):1125-31.
47. Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Molecular Cell. 2000 Jul 1;6(1):77-86.
48. Spiegelman BM, Flier JS. Obesity and the regulation of energy balance. Cell. 2001 Feb 23;104(4):531-43.
49. Guan HP, Chen G. Factors affecting insulin-regulated hepatic gene expression. In: Progress in Molecular Biology and Translational Science 2014 Jan 1 (Vol. 121, pp. 165-215). Academic Press.
50. Rondinone CM. Kinase-dependent pathways and the development of insulin resistance in hepatocytes. Expert Review of Endocrinology & Metabolism. 2007 Mar 1;2(2):195-203.
51. Taskinen MR, Packard CJ, Borén J. Dietary fructose and the metabolic syndrome. Nutrients. 2019 Sep;11(9):1987.
52. Macedo MP, Lima IS, Gaspar JM, Afonso RA, Patarrão RS, Kim YB, et al. Risk of postprandial insulin resistance: the liver/vagus rapport. Reviews in Endocrine and Metabolic Disorders. 2014 Mar 1;15(1):67-77.
53. Ashino NG, Saito KN, Souza FD, Nakutz FS, Roman EA, Velloso LA, et al. Maternal high-fat feeding through pregnancy and lactation predisposes mouse offspring to molecular insulin resistance and fatty liver. The Journal of Nutritional Biochemistry. 2012 Apr 1;23(4):341-8.
54. Ali MA, Eid RM, Hanafi MY. Vitamin C and E chronic supplementation differentially affect hepatic insulin signaling in rats. Life Sciences. 2018 Feb 1;194:196-204.
55. Unger RH. Lipid overload and overflow: metabolic trauma and the metabolic syndrome. Trends in Endocrinology & Metabolism. 2003 Nov 1;14(9):398-403.
56. Arrieta-Cruz I, Su Y, Gutiérrez-Juárez R. Suppression of endogenous glucose production by isoleucine and valine and impact of diet composition. Nutrients. 2016 Feb;8(2):79.
57. Singhal NS, Lazar MA, Ahima RS. Central resistin induces hepatic insulin resistance via neuropeptide Y. Journal of Neuroscience. 2007 Nov 21;27(47):12924-32.
58. Ono H, Pocai A, Wang Y, Sakoda H, Asano T, Backer JM, et al. Activation of hypothalamic S6 kinase mediates diet-induced hepatic insulin resistance in rats. The Journal of Clinical Investigation. 2008 Aug 1;118(8):2959-68.
59. Gislén A, Dacke M, Kröger RH, Abrahamsson M, Nilsson DE, Warrant EJ. Superior underwater vision in a human population of sea gypsies. Current Biology. 2003 May 13;13(10):833-6.
60. Fisher SJ, Kahn CR. Insulin signaling is required for insulin’s direct and indirect action on hepatic glucose production. The Journal of Clinical Investigation. 2003 Feb 15;111(4):463-8.
61. Haas JT, Miao J, Chanda D, Wang Y, Zhao E, Haas ME, et al. Hepatic insulin signaling is required for obesity-dependent expression of SREBP-1c mRNA but not for feeding-dependent expression. Cell Metabolism. 2012 Jun 6;15(6):873-84.
62. Lakshmanan MR, Nepokroeff CM, Porter JW. Control of the synthesis of fatty-acid synthetase in rat liver by insulin, glucagon, and adenosine 3′: 5′ cyclic monophosphate. Proceedings of the National Academy of Sciences. 1972 Dec 1;69(12):3516-9
63. Shimano H, Yahagi N, Amemiya-Kudo M, Hasty AH, Osuga JI, Tamura Y, et al. Sterol regulatory elementbinding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. Journal of Biological Chemistry. 1999 Dec 10;274(50):35832-9.
64. Chen G, Liang G, Ou J, Goldstein JL, Brown MS. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proceedings of the National Academy of Sciences. 2004 Aug 3;101(31):11245-50.
65. Scorletti E, Bhatia L, McCormick KG, Clough GF, Nash K, Hodson L, et al. Effects of purified eicosapentaenoic and docosahexaenoic acids in nonalcoholic fatty liver disease: results from the Welcome* study. Hepatology. 2014 Oct;60(4):1211-21.
66. Zhang Y, Chen W, Li R, Li Y, Ge Y, Chen G. Insulinregulated Srebp-1c and Pck1 mRNA expression in primary hepatocytes from zucker fatty but not lean rats is affected by feeding conditions. PLoS One. 2011 Jun 22;6(6):e21342.
67. Zucker LM, Zucker TF. Fatty, a new mutation in the rat. Journal of Heredity. 1961 Nov 1;52(6):275-8.
68. Iida M, Murakami T, Ishida K, Mizuno A, Kuwajima M, Shima K. Substitution at codon 269 (glutamine→ proline) of the leptin receptor (OB-R) cDNA is the only mutation found in the Zucker fatty (fa/fa) rat. Biochemical and Biophysical Research Communications. 1996 Jul 16;224(2):597-604.
69. Phillips MS, Liu Q, Hammond HA, Dugan V, Hey PJ, Caskey CT, et al. Leptin receptor missense mutation in the fatty Zucker rat. Nature Genetics. 1996 May;13(1):18- 9.
70. Takaya K, Ogawa Y, Isse N, Okazaki T, Satoh N, Masuzaki H, et al. Molecular cloning of rat leptin receptor isoform complementary DNAs—identification of a missense mutation in Zucker fatty (fa/fa) rats. Biochemical and biophysical research communications. 1996 Aug 5;225(1):75-83.
71. Himsworth HP. Dietetic factors influencing the glucose tolerance and the activity of insulin. The Journal of Physiology. 1934 Mar 29;81(1):29.
72. Gloy VL, Briel M, Bhatt DL, Kashyap SR, Schauer PR, Mingrone G, et al. Bariatric surgery versus non-surgical treatment for obesity: a systematic review and metaanalysis of randomised controlled trials. BMJ. 2013 Oct 22;347:f5934.
73. Mingrone G, Panunzi S, De Gaetano A, Guidone C, Iaconelli A, Leccesi L, et al. Bariatric surgery versus conventional medical therapy for type 2 diabetes. New England Journal of Medicine. 2012 Apr 26;366(17):1577- 85.
74. Schauer PR, Kashyap SR, Wolski K, Brethauer SA, Kirwan JP, Pothier CE, et al. Bariatric surgery versus intensive medical therapy in obese patients with diabetes. New England Journal of Medicine. 2012 Apr 26;366(17):1567-76.
75. Mingrone G, Panunzi S, De Gaetano A, Guidone C, Iaconelli A, Nanni G, et al. Bariatric–metabolic surgery versus conventional medical treatment in obese patients with type 2 diabetes: 5 year follow-up of an open-label, single-centre, randomised controlled trial. The Lancet. 2015 Sep 5;386(9997):964-73.
76. Cummings DE, Arterburn DE, Westbrook EO, Kuzma JN, Stewart SD, Chan CP, et al. Gastric bypass surgery vs intensive lifestyle and medical intervention for type 2 diabetes: the CROSSROADS randomised controlled trial. Diabetologia. 2016 May 1;59(5):945-53.
77. Halperin F, Ding SA, Simonson DC, Panosian J, Goebel-Fabbri A, Wewalka M, et al. Roux-en-Y gastric bypass surgery or lifestyle with intensive medical management in patients with type 2 diabetes: feasibility and 1-year results of a randomized clinical trial. JAMA Surgery. 2014 Jul 1;149(7):716-26.
78. Ikramuddin S, Korner J, Lee WJ, Connett JE, Inabnet WB, Billington CJ, et al. Roux-en-Y gastric bypass vs intensive medical management for the control of type 2 diabetes, hypertension, and hyperlipidemia: the Diabetes Surgery Study randomized clinical trial. JAMA. 2013 Jun 5;309(21):2240-9.
79. Courcoulas AP, Goodpaster BH, Eagleton JK, Belle SH, Kalarchian MA, Lang W, et al. Surgical vs medical treatments for type 2 diabetes mellitus: a randomized clinical trial. JAMA Surgery. 2014 Jul 1;149(7):707-15.
80. Courcoulas AP, Belle SH, Neiberg RH, Pierson SK, Eagleton JK, Kalarchian MA, et al. Three-year outcomes of bariatric surgery vs lifestyle intervention for type 2 diabetes mellitus treatment: a randomized clinical trial. JAMA Surgery. 2015 Oct 1;150(10):931-40.
81. Rubino F, Nathan DM, Eckel RH, Schauer PR, Alberti KG, Zimmet PZ, et al. Metabolic surgery in the treatment algorithm for type 2 diabetes: a joint statement by international diabetes organizations. Surgery for Obesity and Related Diseases. 2016 Jul 1;12(6):1144-62.
82. Abdeen G, Le Roux CW. Mechanism underlying the weight loss and complications of Roux-en-Y gastric bypass. Review. Obesity Surgery. 2016 Feb 1;26(2):410- 21.
83. Chen W, Howell ML, Li Y, Li R, Chen G. Vitamin A and feeding statuses modulate the insulin-regulated gene expression in Zucker lean and fatty primary rat hepatocytes. PloS One. 2014 Aug 8;9(8):e100868.
84. Niba ET, Nagaya H, Kanno T, Tsuchiya A, Gotoh A, Tabata C, et al. Crosstalk between PI3 kinase/PDK1/Akt/ Rac1 and Ras/Raf/MEK/ERK pathways downstream PDGF receptor. Cellular Physiology and Biochemistry. 2013;31(6):905-13.
85. Mitsiades CS, Mitsiades N, Koutsilieris M. The Akt pathway: molecular targets for anti-cancer drug development. Current Cancer Drug Targets. 2004 May 1;4(3):235-56.
86. Chen W, Goff MR, Kuang H, Chen G. Higher protein kinase C ? in fatty rat liver and its effect on insulin actions in primary hepatocytes. PloS One. 2015 Mar 30;10(3):e0121890.