Abstract
Epithelial-Mesenchymal Transition (EMT) and its reverse process, Mesenchymal-to-Epithelial Transition (MET) are well-studied processes involved in cell differentiation during development and organ formation, as well as in its reverse process in somatic cell reprogramming. In addition, involvement of a deregulated form of the EMT in tumor progression has long been known. Understanding the molecular and cellular mechanisms for cancer development, progression, and metastasis is a fundamental challenge toward advancing and improving therapeutic strategies. In this mini-review, by considering the distinct phenotypes of available Dido mutations in mice, for the first time we connect Dido gene function and participation with EMT and MET in the areas of cell differentiation, reprogramming, and cancer development.
Keywords
Epithelial-mesenchymal transition, Cell differentiation, Somatic cell reprogramming, Gene expression, Tumor formation, metastasis, Dido gene
The Physiological Occurrence of EMT and MET
The human body is constituted by more than 200 committed cell types [1], all of which derive from the zygote, a single totipotent cell that divides and differentiates into all the cells of an organism throughout its development. Out of this diversity, we focus our interest here on the epithelial and the mesenchymal cells.
Each of these cell types displays specific characteristics. Epithelial cells have apicobasal polarity, are connected by cell junctions, and can form epithelial layers that cover many tissues, whereas mesenchymal cells lack apicobasal polarity, are less connected, but are more motile [2]. Both cell types can nonetheless undergo highly connected mutual transitions. EMT and its reverse process MET are thus well-studied in embryonic development, and several rounds of EMT and MET are described for the generation of various organs throughout their differentiation and development. Molecular studies to analyze these transitions show the involvement of several regulatory networks of transcriptional and translational regulators, linked to post-transcriptional and posttranslational modifications. Gene and protein regulation participate in cellular processes such as cell-cell contact, cell adhesion, cell polarity, cytoskeletal organization (which implies cell migration), all of which are responsible for successful, correct transitions [3-5].
During embryonic development, zygote totipotency is lost after successive differentiation rounds. Using somatic cells, it has recently become possible to develop Induced Pluripotent Cells (iPSC), which have very similar totipotency throughout somatic cell reprogramming [6,7]. Reprogramming is antagonistic to cell differentiation, although both processes share many common players and mechanisms [8]. Overexpression of the so-called Yamanaka factors (Oct4, Klf4, Sox2, and c-Myc) that allow somatic cell reprogramming to iPSC [9,10] was a breakthrough. This finding is important not only for understanding the underlying mechanisms, but also given the potential for regenerative medicine, disease modeling, and disease therapy.
Several studies describe a role for MET transition in early reprogramming phases [11-13], whereas EMT-like changes might be necessary in later reprogramming stages. Results from sequential reprogramming with higher efficiency also postulate an initial EMT-MET that helps erase the cell epigenetic memory and facilitates cell fate conversion [14]. Many of these findings have been described and are discussed extensively in several recent reviews [2,15].
The Dido Gene is Involved in Differentiation and Somatic Cell Reprogramming
The Dido locus is a gene complex that encodes three proteins –Dido1, Dido2, and Dido3– that are generated through alternative splicing. The common N-terminal part of the three isoforms bears a plant homeodomain (PHD) [16,17] that binds to histone 3 trimethylated on lysine 4 (H3K4me3) and connects Dido to chromatin remodeling processes [18]. Dido3 and Dido2 also have a transcription elongation factor S-II subunit M (TFSIIM) domain [17], which mediates binding to RNA polymerase II (RNA pol II) [19,20], and also bear the SPOC domain, linked to Spen family proteins and involved in transcriptional repression [21]. The last Dido exon, exon 16, is specific to the largest isoform, Dido3; it has an arginine-/proline-rich, lowcomplexity region, a central coiled-coil domain, and a C-terminal RNA recognition motif. Dido3 is expressed ubiquitously, whereas the smaller splice variants are detected only transiently.
Deletion of Dido exons 3 and 4 (DidoΔNT) results in N-terminal truncation of all three isoforms. This leads to genomic instability due to centrosome amplification and centromere-localized DNA breaks [22,23], but yields live mice, although they develop myeloid neoplasms [16].
It is of interest to note that Dido3 is also related to stemness, as it is expressed strongly in gastrulating embryos, a time at which EMT is critically important [24], as well as in Embryonic Stem Cells (ESC) and in iPSC. In contrast, it is downregulated during differentiation, and is less expressed in differentiated somatic cells [18]. It is thus not surprising that deletion of Dido3-specific exon 16 results in embryonic lethality at day 8.5. This phenotype is reported for DidoΔCT mutant mice, with constitutive deletion of the C-terminal part (equivalent to the coding part of exon 16) [25], and also for the conditional targeted mutant Dido3ΔE16, with loxP sites flanking exon 16 after Cre recombination [26]. ESC derived from both of these mutants do not differentiate in vitro, but maintain their self-renewal capacity [25,26].
Another notable observation is that Dido3ΔCT/ Dido3ΔE16 ESC are able to self-renew without Leukemia Inhibitory Factor (LIF) supplementation of the culture medium. In these conditions the cells change morphology, grow as a monolayer and are flattened, with a fibroblast-like shape. When injected into nude mice to form teratomas, these tumors are better termed teratocarcinomas; they maintain stem cell marker expression and apparently do not differentiate into cells of the three germ layers (unpublished results, A. Fütterer), in contrast to equivalent ESC cultured with LIF, which maintain typical ESC morphology [25].
Dido3 participates in correct splicing of its own isoforms, as Dido3ΔCT/Dido3ΔE16 mutants do not express the smallest isoform, Dido1 [26]. This is important because Dido1 must be upregulated during differentiation [18] and is involved in downregulation of stemness genes such as Pou5f1 (which encodes Oct4) [20]. As Dido3ΔCT mutants lack the C-terminal binding site, they do not express Dido3 on centrosomes [27]. In addition, interaction of the SFPQ splicing factor with exon 16 links Dido3 to correct, efficient RNA splicing and alternative polyadenylation [28,29]. The connection of Dido3 to cell polarization should also be noted; Dido3ΔCT mutant ESC centrosomes do not position correctly when cell polarization is necessary at the onset of differentiation [20].
We recently found that Dido3 is relevant not only for differentiation, but also for somatic cell reprogramming. For these experiments, we crossed heterozygous Dido3-exon16 floxed mice and transgenic Tg(tetOPou5f1, Sox2,Klf4,Myc)1Srn [7] mice with doxycyclineinducible expression of the reprogramming genes. From the offspring, we derived Dido3ΔE16 Mouse Embryonic Fibroblasts (MEF), which were infected with Ad5CMVCre virus to induce homozygous deletion of exon 16. When treated with doxycycline, these MEF do not undergo reprogramming [26]. In addition, we studied the molecular mechanism and found that Dido3 exon 16 interacts with the helicase Dhx9, whose functions include participation in R-loop processing and in transcription termination [30]. Our analysis of Dido3ΔE16 mutant ESC and MEF using transcriptomic tools, ChIP-seq, RNAseq, protein interaction, and cellular studies identified a central role for Dido3, specifically Dido3 exon16, in regulating RNA metabolism. Exon16 deletion triggered defects in RNA splicing and RNA termination. We verified high-throughput data on some important sample genes such as Carm1, with a role in early developmental differentiation [31], Jam2, associated with high stem cell expression [32] and involvement in cell polarization [33], and Pou5f1/Oct4, the main gatekeeper for self-renewal in ESC [34]. We detected transcriptional read-through provoked by an inability to resolve the increase in R-loop formation, caused by impaired Dhx9 helicase recruitment through Dido3 exon16. Both cell types suffer genomic instability, DNA damage, and replication stress; ESC thus do not differentiate nor do MEF undergo somatic cell reprogramming [26].
The phenotype of the DidoΔNT mutant MEF differs remarkably from that of exon16-deleted mutants. Although this mutant has RNA splicing defects [28], there is no increase in R-loops and replication stress; ESC derived from these mice can differentiate in vitro, and the MEF undergo somatic reprogramming [26].
These data led us to test whether the genes whose expression is altered by exon16 deletion were identified in reprogramming studies. We combined our gene expression data from Dido3ΔE16 ESC in self-renewal conditions and from embryonic bodies (EB) at the onset of differentiation (GEO: GSE85029) [20] and GEO: GSE152346 [26]) with those from Samavarchi-Tehrani et al. [12]. The latter study, an exhaustive time-course microarray analysis of the reprogramming process, found the Dido gene in the initiation phase, at the time MET is activated (Figure 1). Encouraged by this result, we identified additional common genes (Table 1). Although we did not detect the so-called classical EMT driver genes [3,35,36], we found several genes necessary in the transition pathways. We divided them into various functional categories such as stemnessrelated, transcription factors, and zinc-finger proteins, genes involved in epigenetic changes (transcriptional and translational), genes related to cell cycle, cytoskeleton, apoptosis and repair, genes coding for enzymes such as ligases or kinases, and other functions (Table 1). This set of genes shows enormous potential to influence the EMT/ MET pathway; further studies are needed to delineate its specific roles.
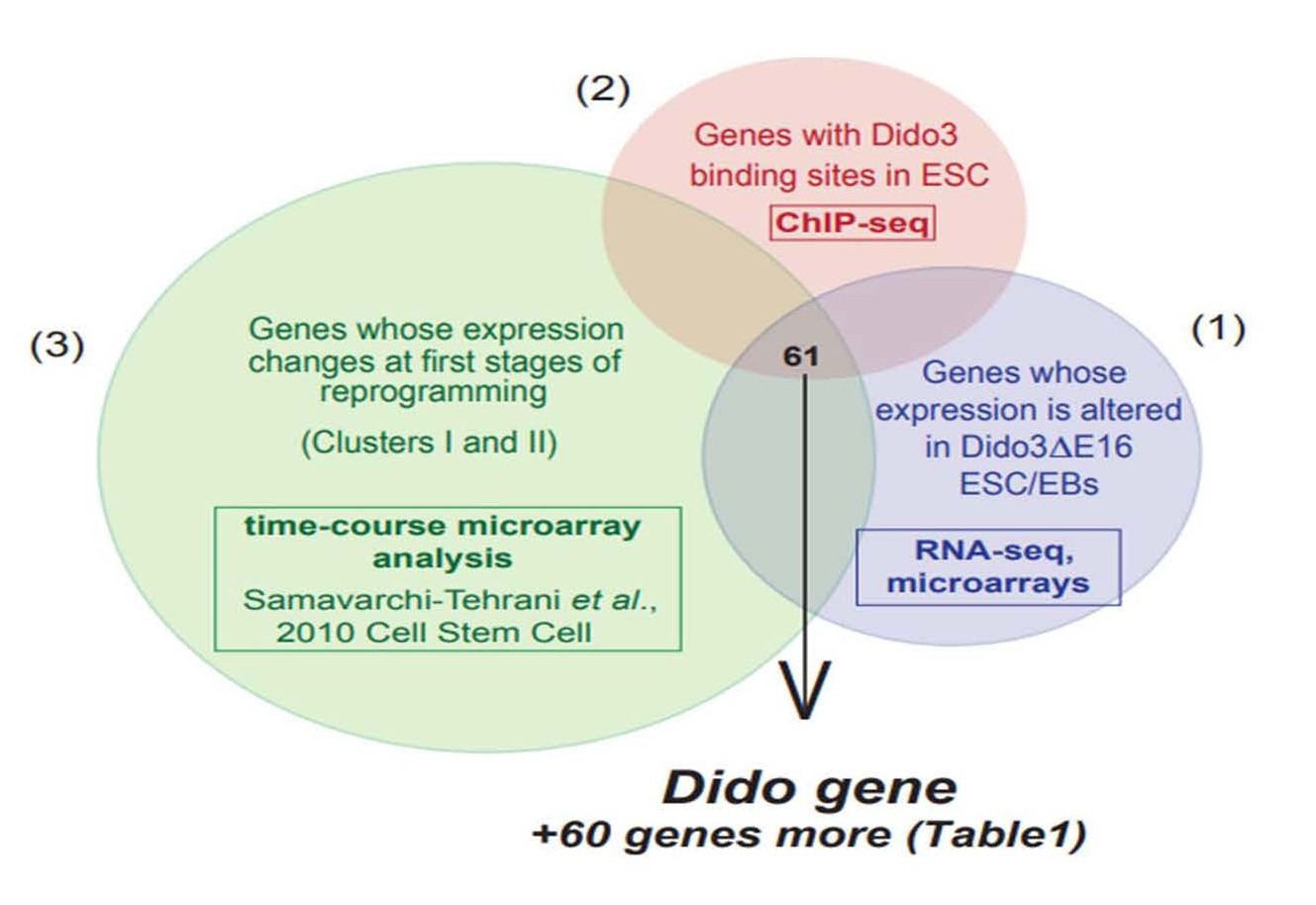
Figure 1. The proportional Venn diagram combines three data sets: (1) genes with altered expression in Dido3ΔE16 ESC and EB compared to wt ESC and EB, respectively (microarray and RNA-seq data), (2) Dido3 DNA binding sites in ESC (ChIP-seq), (3) published data: a compendium of genes whose expression changes at early stages of MEF reprogramming (cluster I and II) (microarray analysis, [12]).
| Stemness related genes | Transcription factor, Zinc-finger |
Epigenetic transcription, translation | Cell cycle, Cytoskeleton, Apoptosis, Repair | Enzymes: Ligase, Kinase |
other |
| Pou5f1 | Klf3 | Dnmt3b | Diablo | Manba | Gtsf1l |
| Esrrb | Pml | Wdr5 | Mybl2 | Pus3 | Ehd3 |
| Zfp42 | Tcf19 | Suz12 | Msh6 | Rnf40 | Spns2 |
| Nr0b1 | Gli1 | Eif2s2 | Kank3 | Mkrn1 | Slc16a3 |
| Tdgf1 | Zfp57 | Phc1 | Cep68 | Tdh | Nvl |
| Fgf4 | Zfp296 | E130012A19Rik | Pdzd4 | Tbl3 | |
| Jam2 | Zfp386 | Ssbp4 | Pmm1 | Dbp | |
| Zfp444 | Psip1 | Apobec3 | Pltp | ||
| Zfp459 | Impa2 | Nup43 | |||
| Zfp710 | Upp1 | ||||
| Zscan22 | Fkbp5 | ||||
| M6pr | |||||
| B4galt6 | |||||
| Got1 | |||||
| Pdk1 | |||||
| Fads3 | |||||
| Ulk1 | |||||
| Neurl4 | |||||
| Gpi1 |
EMT/MET in Tumor Formation and Cancer Progression
EMT and MET proceed not only in physiological conditions, but also in a pathological manner, and deregulated EMT participates in tumor formation and cancer metastasis by providing cells with migratory and invasive properties [3,5,36-38]. Note that EMT in cancer is mostly partial and does not complete the transition [39-42]. Many tumor cells remain in transition states or undergo dynamic, reversible transitions and can express epithelial and mesenchymal characteristics, which is linked to increased tumor aggressiveness [43]. On the other hand, MET in circulating cancer cells can lead to secondary tumors when they invade a metastatic niche [42,44].
As stated above, Dido expression changes do not affect the classical EMT-TF (transcription factors) that drive EMT or MET initiation. As also mentioned and reviewed in depth by others [45,46], cancer initiation and progression depend on altered regulation at several levels, including transcriptional and translational processes, expression of non-coding RNA, alternative splicing events, and protein stability; Dido is likely to play its important role in these areas. Evidence for this hypothesis was demonstrated by links of Dido function to alternative splicing, transcription termination, and protein stability [26,28,29]. Another central feature in the control of EMT derives from EMTTF interaction with epigenetic modifiers and modulators of chromatin configuration [38,39,45,46]. Epigenetic shifts enable cell plasticity. Candidate genes such as Wdr5 (WD repeat-containing protein 5), Dnmts (DNA methyltransferases), and the methyltransferase Suz12 (part of the Polycomb 2 repressor complex) (Table 1), all of which are influenced by Dido, have also been discussed as possible epigenetic modification components in EMT [45,46]. The significance of Dido in this field was recently underlined by an exhaustive computational study that integrated multi-omics data and Dido domain composition, to propose a molecular mechanism for Dido3 in RNA pol II pausing and long-range chromatin interactions, which have pleiotropic consequences [47].
Future Perspectives
Our description of Dido gene involvement in hematological myeloid neoplasms [16] and the work of several other groups implicating Dido in tumor progression or prognosis (with some contradictory results) [48-51] identify an avenue for further research. The contrasting results in cancer studies might derive from the fact that most did not distinguish between different Dido isoforms and their autoregulation.
Another question regarding Dido influence in cancer arises from the cell reprogramming results. Considering the failure of primary Dido3ΔE16 MEF in somatic cell reprogramming, one might anticipate reduced tumor transformation capacity. The point of interest here is that immortalization of these MEF not only restores, but also greatly increases their reprogramming capacity [26]. Extrapolating these results and bearing in mind that inactivation of p53 and/or RB (retinoblastoma) pathways in cancer is a common phenomenon, it will be very interesting to determine the outcome of additional Dido mutations in cancer cells, and whether Dido has a role in cancer stem cell development.
A simplified scheme illustrates EMT and MET processes in differentiation, cell reprogramming, and tumor formation, and the likely involvement of the Dido gene (Figure 2). The need to understand Dido gene function in tumor formation and metastasis encourages us to explore and clarify its role in oncogenic transformation and cancer progression, further instances in which EMT and MET are fundamental.
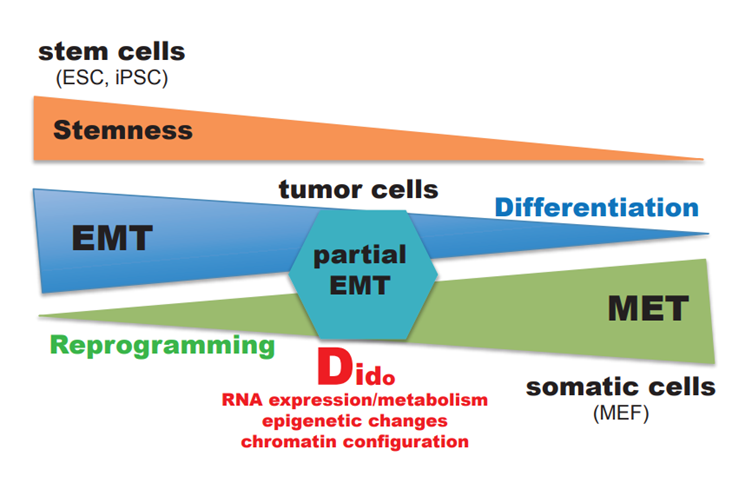
Figure 2. Simplified model of the working hypothesis, with possible connections and influence of the Dido gene to EMT and MET processes.
Funding
This work was supported by grants SAF2016-75456R and PID2019-110574RB-I00 from the Spanish Ministerio de Ciencia e Innovación and from the Comunidad de Madrid (MITIC), and from CSIC Intramural Project PIE 201920E017.
Conflict of Interest
The authors declare no conflict of interests. The authors thank Catherine Mark for editorial assistance.
References
2. Shu X, Pei D. The function and regulation of mesenchymal-to-epithelial transition in somatic cell reprogramming. Current Opinion in Genetics & Development. 2014 Oct 1;28:32-7.
3. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelialmesenchymal transitions in development and disease. Cell. 2009 Nov 25;139(5):871-90.
4. Chen T, You Y, Jiang H, Wang ZZ. Epithelial– mesenchymal transition (EMT): a biological process in the development, stem cell differentiation, and tumorigenesis. Journal of Cellular Physiology. 2017 Dec;232(12):3261-72.
5. Kim DH, Xing T, Yang Z, Dudek R, Lu Q, Chen YH. Epithelial mesenchymal transition in embryonic development, tissue repair and cancer: a comprehensive overview. Journal of Clinical Medicine. 2018 Jan;7(1):1.
6. Ohnuki M, Takahashi K. Present and future challenges of induced pluripotent stem cells. Philosophical Transactions of the Royal Society B: Biological Sciences. 2015 Oct 19;370(1680):20140367.
7. Abad M, Mosteiro L, Pantoja C, Cañamero M, Rayon T, Ors I, et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature. 2013 Oct;502(7471):340-5.
8. Takahashi K, Yamanaka S. A developmental framework for induced pluripotency. Development. 2015 Oct 1;142(19):3274-85.
9. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663-76.
10. Yamanaka S. Pluripotency and nuclear reprogramming. Philos Trans R Soc Lond B Biol Sci. 2008;363(1500):2079-87.
11. Li R, Liang J, Ni S, Zhou T, Qing X, Li H, et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell. 2010;7(1):51-63.
12. Samavarchi-Tehrani P, Golipour A, David L, Sung HK, Beyer TA, Datti A, et al. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell. 2010;7(1):64-77.
13. Hansson J, Rafiee Mahmoud R, Reiland S, Polo Jose M, Gehring J, Okawa S, et al. Highly Coordinated Proteome Dynamics during Reprogramming of Somatic Cells to Pluripotency. Cell Reports. 2012;2(6):1579-92.
14. Liu X, Sun H, Qi J, Wang L, He S, Liu J, et al. Sequential introduction of reprogramming factors reveals a time-sensitive requirement for individual factors and a sequential EMT–MET mechanism for optimal reprogramming. Nature Cell Biology. 2013;15(7):829-38.
15. Pei D, Shu X, Gassama-Diagne A, Thiery JP. Mesenchymal–epithelial transition in development and reprogramming. Nature Cell Biology. 2019 Jan;21(1):44-53.
16. Fütterer A, Campanero MR, Leonardo E, Criado LM, Flores JM, Hernández JM, et al. Dido gene expression alterations are implicated in the induction of hematological myeloid neoplasms. The Journal of Clinical Investigation. 2005 Sep 1;115(9):2351-62.
17. Rojas AM, Sanchez-Pulido L, Fütterer A, Van Wely KH, Martinez-A C, Valencia A. Death inducer obliterator protein 1 in the context of DNA regulation: Sequence analyses of distant homologues point to a novel functional role. The FEBS Journal. 2005 Jul;272(14):3505-11.
18. Gatchalian J, Fütterer A, Rothbart SB, Tong Q, Rincon-Arano H, de Diego AS, et al. Dido3 PHD modulates cell differentiation and division. Cell Reports. 2013 Jul 11;4(1):148-58.
19. Kinkelin K, Wozniak GG, Rothbart SB, Lidschreiber M, Strahl BD, Cramer P. Structures of RNA polymerase II complexes with Bye1, a chromatin-binding PHF3/DIDO homologue. Proceedings of the National Academy of Sciences. 2013 Sep 17;110(38):15277-82.
20. Fütterer A, de Celis J, Navajas R, Almonacid L, Gutiérrez J, Talavera-Gutiérrez A, et al. DIDO as a switchboard that regulates self-renewal and differentiation in embryonic stem cells. Stem Cell Reports. 2017 Apr 11;8(4):1062-75.
21. Sánchez-Pulido L, Rojas AM, Van Wely KH, Martinez-A C, Valencia A. SPOC: a widely distributed domain associated with cancer, apoptosis and transcription. BMC Bioinformatics. 2004 Dec;5(1):1-6.
22. Guerrero AA, Gamero MC, Trachana V, Fütterer A, Pacios-Bras C, Díaz-Concha NP, et al. Centromerelocalized breaks indicate the generation of DNA damage by the mitotic spindle. Proceedings of the National Academy of Sciences. 2010 Mar 2;107(9):4159-64.
23. Trachana V, van Wely KH, Guerrero AA, Fütterer A, Martínez-A C. Dido disruption leads to centrosome amplification and mitotic checkpoint defects compromising chromosome stability. Proceedings of the National Academy of Sciences. 2007 Feb 20;104(8):2691-6.
24. Nakaya Y, Sheng G. Epithelial to mesenchymal transition during gastrulation: An embryological view. Development, Growth & Differentiation. 2008;50(9):755-66.
25. Fütterer A, Raya Á, Llorente M, Izpisúa-Belmonte JC, De La Pompa JL, Klatt P, et al. Ablation of Dido3 compromises lineage commitment of stem cells in vitro and during early embryonic development. Cell Death & Differentiation. 2012 Jan;19(1):132-43.
26. Fütterer A, Talavera-Gutiérrez A, Pons T, de Celis J, Gutiérrez J, Plaza VD, Martínez-A C. Impaired stem cell differentiation and somatic cell reprogramming in DIDO3 mutants with altered RNA processing and increased R-loop levels. Cell Death & Disease. 2021 Jun 21;12(7):1-4.
27. De Diego AS, Guerrero AA, Martínez-a C, Van Wely KH. Dido3-dependent HDAC6 targeting controls cilium size. Nature Communications. 2014 Mar 25;5(1):1-1.
28. Mora Gallardo C, Sánchez de Diego A, Gutiérrez Hernández J, Talavera-Gutiérrez A, Fischer T, Martínez-A C, van Wely KH. Dido3-dependent SFPQ recruitment maintains efficiency in mammalian alternative splicing. Nucleic Acids Research. 2019 Jun 4;47(10):5381-94.
29. Mora Gallardo C, Sánchez de Diego A, Martínez-A C, van Wely KHM. Interplay between splicing and transcriptional pausing exerts genome-wide control over alternative polyadenylation. Transcription. 2021:1-17.
30. Cristini A, Groh M, Kristiansen MS, Gromak N. RNA/DNA hybrid interactome identifies DXH9 as a molecular player in transcriptional termination and R-loop-associated DNA damage. Cell Reports. 2018 May 8;23(6):1891-905.
31. Bedzhov I, Graham SJ, Leung CY, Zernicka-Goetz M. Developmental plasticity, cell fate specification and morphogenesis in the early mouse embryo. Philosophical Transactions of the Royal Society B: Biological Sciences. 2014 Dec 5;369(1657):20130538.
32. Sakaguchi T, Nishimoto M, Miyagi S, Iwama A, Morita Y, Iwamori N, Nakauchi H, Kiyonari H, Muramatsu M, Okuda A. Putative “stemness” gene jam-B is not required for maintenance of stem cell state in embryonic, neural, or hematopoietic stem cells. Molecular and Cellular Biology. 2006 Sep 1;26(17):6557-70.
33. Matter K, Aijaz S, Tsapara A, Balda MS. Mammalian tight junctions in the regulation of epithelial differentiation and proliferation. Current Opinion in Cell Biology. 2005 Oct 1;17(5):453-8.
34. Pesce M, Schöler HR. Oct-4: gatekeeper in the beginnings of mammalian development. Stem Cells. 2001 Jul;19(4):271-8.
35. Kim HY, Jackson TR, Davidson LA. On the role of mechanics in driving mesenchymal-to-epithelial transitions. Seminars in Cell & Developmental Biology. 2017 Jul 1;67:113-122.
36. Lai X, Li Q, Wu F, Lin J, Chen J, Zheng H, Guo L. Epithelial-Mesenchymal Transition and Metabolic Switching in Cancer: Lessons From Somatic Cell Reprogramming. Frontiers in Cell and Developmental Biology. 2020 Aug 6;8:760.
37. Polo JM, Hochedlinger K. When fibroblasts MET iPSCs. Cell stem cell. 2010 Jul 2;7(1):5-6.
38. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nature reviews Molecular Cell Biology. 2019 Feb;20(2):69-84.
39. Roche J. Erratum: Roche, J. The Epithelial-to- Mesenchymal Transition in Cancer. Cancers, 2018, 10, 52. Cancers. 2018 Mar;10(3):79.
40. Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nature Reviews Cancer. 2018 Feb;18(2):128-34.
41. Lu W, Kang Y. Epithelial-mesenchymal plasticity in cancer progression and metastasis. Developmental Cell. 2019 May 6;49(3):361-74.
42. Ribatti D, Tamma R, Annese T. Epithelialmesenchymal transition in cancer: a historical overview. Translational Oncology. 2020 Jun 1;13(6):100773.
43. Jolly MK, Boareto M, Huang B, Jia D, Lu M, Ben- Jacob E, Onuchic JN, Levine H. Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Frontiers in Oncology. 2015 Jul 20;5:155.
44. Banyard J, Bielenberg DR. The role of EMT and MET in cancer dissemination. Connective Tissue Research. 2015 Sep 3;56(5):403-13.
45. De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nature Reviews Cancer. 2013 Feb;13(2):97-110.
46. Tam WL, Weinberg RA. The epigenetics of epithelialmesenchymal plasticity in cancer. Nature Medicine. 2013;19(11):1438-49.
47. Pons T, Serra F, Pazos F, Valencia A, Martínez-A C. DIDO3 acts at the interface of RNAPII transcription and chromatin structure regulation. 2021. bioRxiv preprint. doi: https://doi.org/10.1101/2021.09.27.462041.
48. Braig S, Bosserhoff AK. Death inducer-obliterator 1 (Dido1) is a BMP target gene and promotes BMP-induced melanoma progression. Oncogene. 2012;32(7):837-48.
49. Forghanifard MM, Naeimi Khorasanizadeh P, Abbaszadegan MR, Javdani Mallak A, Moghbeli M. Role of DIDO1 in Progression of Esophageal Squamous Cell Carcinoma. Journal of Gastrointestinal Cancer. 2019;51(1):83-7.
50. Zhang Y, Jiang J, Zhang J, Shen H, Wang M, Guo Z, et al. CircDIDO1 inhibits gastric cancer progression by encoding a novel DIDO1-529aa protein and regulating PRDX2 protein stability. Molecular Cancer. 2021 Dec;20(1):1-7.
51. Li J, Wang AS, Wang S, Wang CY, Xue S, Li WY, et al. Death-inducer obliterator 1 (DIDO1) silencing suppresses the growth of bladder cancer cells through decreasing SAPK/JNK signaling cascades. Neoplasma. 2020 Jan 1;67(5):1074-84.