Abstract
The MYCN oncoprotein has been notoriously undruggable and is infamous for causing aggressive cancer with poor outcomes in children and adults. Following surgery, radiation, and chemotherapy, patients who develop progressive disease have few treatment options. An analysis of the dysregulated protein network caused by MYCN amplification suggested co-targeting PLK1, AURKA, CKS1, AKT, MTOR, and USP7 would be useful to take advantage of synthetic lethal vulnerabilities while overcoming redundancies and resistance mechanisms that stabilize N-Myc by preventing its proteasome degradation. Naturopathic compounds, (genistein, tanshinone, resveratrol, betulinic acid) and fluoxetine were re-purposed to target the complex protein network in a patient with MYCN-amplified and PTEN-deficient multifocal, relapsed anaplastic ependymoma following standard therapy. The patient achieved a clinically meaningful and durable response for 6 months prior to developing disease progression characterized by chromosome 11q (YAP1, BIRC2/3) amplification. The experience suggests molecularly-informed integration of naturopathic compounds can have utility for disease control and survival. The success, although anecdotal, suggests that the previous failure of single agent strategies could be overcome with a network targeting approach that simultaneously precipitates cell cycle arrest, rescues FBXW7 ubiquitination, and enhances oxidative stress. As such, MYCN may no longer be strictly unactionable but appears amenable to co-targeting key nodes in its self-sustaining disease network.
Keywords
Anaplastic ependymoma, MYCN, PLK1, AURKA, AKT, MTOR, FBXW7, USP7, miR-34a, Signaling pathway, Network targeting, Synthetic lethality, Integrative oncology
Background
Ependymoma is a rare form of glioma arising from the ependymal cells that line the ventricular surfaces of the brain and spinal canal. Different forms of the disease arise in adults and children and are classified by site of origin (e.g., supratentorial (ST), posterior fossa (PF), spinal cord (SC)), by histologic grade (WHO CNS Grade 2 or 3), and molecular drivers. Surgical resection followed by radiation therapy represents standard treatment for newly diagnosed patients. However, such rare cancers present a challenge as few if any randomized trials are available to inform care. As such, no standard of care existed for recurrent ependymoma until the recent phase II study of temozolomide and lapatinib showed antitumor benefit with some complete responses, a median progression free survival of 7.8 months, and decrease in symptom burden [1]. However, not all patients benefit from this approach and even those who respond eventually relapse and are in need of novel treatment approaches.
An especially aggressive and poor prognosis form of adult-onset anaplastic ependymoma is driven by N-myc proto-oncogene (MYCN) amplification [2,3]. The MYCN gene codes for the protein N-Myc, an E-box binding, basic helix leucine zipper (bHLH-LZ) transcription factor which orchestrates a network of signaling pathway aberrations characterized by over-expression of key oncogenes (e.g., mouse double minute 2 homolog E3 ubiquitin-protein ligase (MDM2), aurora kinase A (AURKA), polo-like kinase-1 (PLK1), and forkhead box protein M1 (FOXM1) and takedown of a key tumor suppressors (e.g., Cellular tumor antigen p53 (p53) and F-box/WD repeat-containing protein 7 (FBXW7)) that together spawn tumorigenesis, aggressive disease behavior, and unsatisfactory treatment outcomes for many different kinds of pediatric and adult cancer.
N-Myc forms a series of redundant positive feedback loops [4]. By inducing transcription of FOXM1, AURKA, and PLK1 N-Myc propels movement through the cell cycle, including G1 → S phase [5,6], the G2/M checkpoint [7], and M phase to cause uncontrolled proliferation and replication stress. FOXM1 drives transcription of AURKA [8] and PLK1 [9], while PLK1 reciprocally phosphorylates FOXM1 to produce hyperactivation [10] Another feed forward loop emerges from the upregulation of MDM2 transcription by N-Myc to promote p53 turnover [11,12]. Enhanced p53 turnover compromises transcription of the master tumor suppressor miR-34a [13] causing increased translation of MYCN and numerous oncogenes [14,15]. Turnover of p53 also enhances PLK1 transcription [16,17] and phosphorylation of p53 by PLK1 inhibits the tumor suppressor function [18]. MDM2 upregulates MYCN mRNA [19,20], as well as activating deubiquitinase enzymes that reverse the ubiquitination of N-Myc [21].
Both AURKA and PLK1 inhibit the function of the critical tumor suppressor FBXW7 [22,23], an E3 ubiquitin kinase that promotes 26S proteasomal turnover of N-Myc, as well as other targets, including key drivers of the malignant phenotype, such as induced myeloid leukemia cell differentiation protein (MCL1), CCNE1/2, c-MYC, neurogenic locus notch homolog protein 1 and 2 (NOTCH1/2), transcription factor Jun (JUN), Krueppel-like factor 5 (KLF5), nuclear factor erythroid 2-related factor 2 protein (NRF2), Rapamycin-insensitive companion of mammalian target of rapamycin (RICTOR), and mammalian Target Of Rapamicin (MTOR) [24]. FOXM1 is also a target of FBXW7 which requires glycogen synthase kinase beta (GSK3-ß) phosphorylation for turnover [25,26]. FOXM1 is responsible upregulation of DNA repair and other components of the therapy-resistant malignant phenotype [27]. Co-existing dysregulation of GSK3-ß and protein phosphatase 2 (PP2A) solidify the disease network further. Upstream phosphoinositide-3 kinase (PI3K)- protein kinase B (PKB) gene (AKT) and MTOR signaling pathways aberrations interfere with phosphorylation of the CDC4 phosphodegron (CPD) targets and impair recognition and turnover of N-Myc and other FBXW7 targets. Effectively, numerous redundant feedforward loops are contained in the protein network that stabilize N-Myc by preventing its degradation and amplifying its transcriptional program to cause a tumorigenic proteome by inhibiting the ubiquitin proteosome system [28] (Table 1).
|
PHENOTYPE |
SIGNALING PATHWAY |
REFERENCES |
|---|---|---|
|
Proliferation |
N-Myc → PLK1 → M_Phase |
77 |
|
N-Myc → AURKA → PLK1 → p21↓ → CDK2_CCNE↓ → S_Phase |
78,79 |
|
|
N-Myc → AURKA → PLK1 → CDC25 → [MYT1 and WEE1] ↓ → CDK1_CCNB1 →[G2/M, M Phase progression] |
80 |
|
|
N-Myc → CSK1_SKP2 → [p27] ↓ → CDK1_CCNA → M_Phase |
81 |
|
|
N-Myc → AURKA → FBXW7↓ → CCNE1↓ → S_Phase |
82 |
|
|
N-Myc → AURKA → PLK1→ FBXW7↓→ CCNE1↓ → S_Phase |
||
|
PTEN → PIP3 → AKT → GSK3ß↓ → FBXW7 → CCNE1↓ → S_Phase |
||
|
N-Myc → AURKA → FBXW7↓ → NOTCH1 |
83 |
|
|
N-Myc → AURKA → FBXW7↓→ KLF5↓ |
84,85 |
|
|
N-Myc → AURKA → PLK1 → FBXW7↓ → KLF5↓ |
||
|
N-Myc → AURKA → FBXW7↓ → RICTOR↓ → OGT (O-GlcNAcylation) → [PI3K, PDK1, AKT, YAP1] → [Proliferation, Survival] |
86 |
|
|
N-Myc → AURKA → PLK1 → FBXW7↓ → RICTOR↓ → OGT (O-GlcNAcylation) → [PI3K, PDK1, AKT, YAP1] → [Proliferation, Survival] |
||
|
N-Myc → AURKA → FBXW7↓ → [JUN, PSEN1]↓ |
87,88 |
|
|
N-Myc → AURKA → PLK1 → FBXW7↓→ [JUN, PSEN1]↓ |
||
|
N-Myc → MDM2 → p53↓→ PLK1↓→ Tumorigenesis |
89 |
|
|
N-Myc → MDM2 → p53↓→ miR-34a → [MYCN, MDM4, BIRC5, MET, CREB, AXL, BCL2, NOTCH1, IL6R, MMP2/9, HOTAIR, SNAL, SLUG, ZEB1, MYC, AVIL, E2F1, CDK6]↓ → Tumorigenesis |
90,91 |
|
|
PTEN → PIP3 → AKT → MDM2 → PLK1 → Tumorigenesis |
|
|
|
PTEN → PIP3 → AKT → GSK3-ß↓ → ßTrCP → ß-catenin↓ → Tumorigenesis |
92 |
|
|
PTEN → PIP3 → AKT → GSK3-ß↓ → ßTrCP → YAP1↓ → [MYC, AREG, CTGF] → Tumorigenesis |
|
|
|
PTEN → PIP3 → AKT → GSK3-ß↓ → ßTrCP → DEPTOR↓ → MTOR↓ → [S6K, 4EBP-1, ATF4, HIF1A] → [Biosynthesis, Translation, Proliferation] |
93,94 |
|
|
Radiation Chemotherapy Resistance |
N-Myc → AURKA → FBXW7↓ → NFE2L2↓ → [GPX4, FTH1] → ROS↓ → Ferroptosis |
95 |
|
N-Myc → AURKA → PLK1 → FBXW7↓→ NFE2L2↓ → [GPX4, FTH1] → ROS↓ → Ferroptosis |
||
|
PTEN → PIP3↓→ AKT → GSK3ß↓→ NFE2L2↓ → ßTrCP |
96,97 |
|
|
AURKA → PLK1 → MITOTIC_CATASTROPHE↓→ Resistance to tubulin targeting strategies |
98,99 |
|
|
AURKAIP1 → AURKA↓→ PLK1 → MITOTIC_CATASTROPHE↓ → Resistance to tubulin targeting strategies |
||
|
PTEN → PIP3↓→ AKT → GSK3ß↓→ FBXW7 → FOXM1↓ → [BRCA2, RAD51] (HRR) → DNA Repair |
100-102 |
|
|
N-Myc → AURKA → FBXW7↓→ FOXM1↓→ [BRCA2, RAD51] (HRR) → DNA Repair |
||
|
N-Myc → AURKA → PLK1 → FBXW7↓ → FOXM1↓ → [BRCA2, RAD51] (HRR) → DNA Repair |
||
|
N-Myc → AURKA → PLK1 → NEK2 → GLI → MGMT → [Temozolomide, CCNU, BCNU, dacarbazine, procarbazine]_Resistance |
103 |
|
|
PTEN → PIP3 → AKT → GSK3-ß↓ → ßTrCP → YAP1↓ → ABCG2 → Multidrug Resistance |
104,105 |
|
|
Cancer Stem Cells |
N-Myc → AURKA → FBXW7↓ → NOTCH1↓ → CSC → Tumor_Re-population |
106,107 |
|
N-Myc → AURKA → PLK1 → FBXW7↓→ NOTCH1/2↓→ CSC → Tumor_Re-population |
||
|
N-Myc → AURKA → PLK1 → CRAF → MEK1/2 → ERK1/2 → ZEB1 → CSC → Tumor_Re-population |
|
|
|
MYCN Stabilization |
N-Myc → AURKA → FBXW7↓→ N-Myc↓ |
108-110 |
|
PTEN → PIP3 → AKT → GSK3ß↓ → FBXW7→ N-Myc↓ |
||
|
CCNB1 → N-Myc_S62 → FBXW7 → N-Myc↓ |
||
|
N-Myc → AURKA → PLK1 → FBXW7↓→ N-Myc↓ |
111-113 |
|
|
N-Myc → AURKA → PLK1 → FBXW7↓→ RICTOR↓ → AKT → GSK3ß↓ → FBXW7→ N-Myc↓ |
114,115 |
|
|
N-Myc → AURKA → PLK1 → FOXM1 → AURKA → FBXW7↓→ N-Myc↓ |
116 |
|
|
|
|
|
|
PTEN → PIP3 → AKT → GSK3ß↓ → N-Myc_T58 → FBXW7 → N-Myc↓ |
|
|
|
PTEN → PIP3 → AKT → GSK3ß↓ → FBXW7 → MTOR↓ → PP2A → N-Myc_S62↓ → N-Myc |
117-120 |
|
|
PTEN → PIP3 → AKT → TSC1/2 ↓ → RHEB → MTOR → PP2A → N-Myc_S62↓ → N-Myc |
||
|
N-Myc → AURKA → FBXW7↓ → MTOR↓ → PP2A → N-Myc_S62 → N-Myc |
||
|
N-Myc → AURKA → PLK1 → FBXW7↓ → MTOR↓→ PP2A → N-Myc_S62↓ → N-Myc |
||
|
N-Myc → MDM2 → N-Myc |
121,122 |
|
|
N-Myc → MDM2 → USP7 → N-Myc |
123 |
|
|
N-Myc → MDM2 → p53↓→ miR-34a →N-Myc↓ |
124-126 |
|
|
N-Myc → MDM2 → XIAP → TGFBR1_TGFBR2 → RHOA → FAK (PTK2) → SRC → AURKA → FBXW7↓ → N-Myc↓ |
127-129 |
|
|
N-Myc → MDM2 → XIAP → TGFBR1_TGFBR2 → RHOA → FAK (PTK2) → SRC → AURKA → PLK1 → FBXW7↓ → N-Myc↓ |
||
|
PLK1 → MDM2 → p53↓ |
130 |
|
|
Apoptotic Blockade |
N-Myc → AURKA → FBXW7↓ → MCL1↓ → Apoptosis↓ |
131 |
|
PTEN → PIP3 → AKT → GSK3ß → FBXW7 → MCL1↓ → Apoptosis↓ |
||
|
N-Myc → AURKA → PLK1 → FBXW7↓ → MCL1↓ → Apoptosis↓ |
||
|
N-Myc → MDM2 → p53↓ → Apoptosis |
132,133 |
|
|
PTEN → PIP3 → AKT → GSK3-ß↓ → ßTrCP → YAP1↓ → BIRC5 → Apoptosis↓ |
134 |
|
|
Angiogenesis |
N-Myc → AURKA → FBXW7↓ → MTOR↓ → HIF1A → VEGF |
135,136 |
|
N-Myc → AURKA → PLK1→ FBXW7↓→ MTOR↓ → HIF1A → VEGF |
||
|
PTEN → PIP3↓→ AKT → GSK3ß↓→ FBXW7 → MTOR↓ → HIF1A → VEGF |
||
|
PTEN → PIP3↓ → AKT → TSC1/2↓ → RHEB → MTOR → HIF1A → VEGF |
||
|
N-Myc → AURKA → FBXW7↓ → MTOR↓ → HIF1A → VEGF |
||
|
N-Myc → AURKA → PLK1→ FBXW7↓→ MTOR↓→ HIF1A → VEGF |
||
|
PTEN → PIP3↓ → AKT → GSK3ß↓ → FBXW7 → MTOR↓ → HIF1A → VEGF |
For the most part, drugs have not been developed to inhibit transcription factors, including the MYC family oncogenes which remain undruggable. However, oncogene amplification causes exquisite sensitivity to the takedown of key proteins, referred to as synthetic lethal partners (SLP) [29]. In MYCN-driven cancers, cyclin kinase subunit-1 (CKS1) [30], polo-like kinase-1 (PLK1), aurora kinase A (AURKA) [31], aurora kinase B (AURKB) [32], and poly-ADP ribose phosphorylase (PARP) [33] are recognized SLP of the MYC family oncogenes. While no PLK1 or AURKA inhibitors have yet received regulatory approval, genistein, a soy isoflavone, has been studied in cancer prevention clinical trials [34] and inhibits PLK1 with a Kd of 7.9 µM [35], a concentration within reach of oral supplementation [6]. Genistein also upregulates miR-34a to reinstate translational blockade of MYCN [37,38]. Tanshinone derived from the rhizome of the Chinese herb Salvia maltorrhiza (danshen) is part of anti-cancer treatment in Traditional Chinese Medicine (TCM) [39] and inactivates AURKA translation by upregulating miR-32 [40] and Let-7a-5p [41]. N-Myc drives the upregulation of casein kinase-1 (CSK1) which promotes the ubiquitin turnover of cyclin-dependent kinase inhibitor 1 protein (p21 - CDKN1A) and cyclin-dependent kinase inhibitor 1B protein (p27 - CDKN1B) the primary brakes on CDK2_CCNE driver of G1 → S phase progression in the cell cycle [42]. Fluoxetine (Prozac™) has been repurposed to block CKS1 in MYCN-driven neuroblastoma to precipitate cell cycle arrest. Resveratrol, a phytoalexin found in many plant species, has been utilized to target AKT, thereby reversing GSK3-ß inactivation as well as activating 5' adenosine monophosphate-activated protein kinase (AMPK) through a phosphodiesterase-4 (PDE4)-dependent mechanism to inhibit MTOR signaling [43,44]. Resveratrol also suppresses the expression of oncogenic micro-RNA 21(miR-21) known to promote expression of B-cell lymphoma 2 (BCL2) [45]. Lastly, the deubiquitinase enzymes also stabilize N-Myc by opposing its proteasomal fate in high grade neuroendocrine cancers [46]. Betulinic acid possesses broad activity to inhibit multiple ubiquitin-specific-processing protease (USP) deubiquitinases for this purpose [47,48].
In principle, overcoming cancer is an engineering problem whose success depends upon deciphering and targeting its underlying complex adaptive network. To embrace this complexity, co-targeting PLK1, AURKA, CKS1, AKT, MTOR, and USP7 could potentially dismantle network redundancies, precipitate proliferation arrest, and rescue FBXW7 to re-establish the normal turnover of N-Myc. A network-targeting strategy to attack these key nodes simultaneously was developed by combining genistein, tanshinone, resveratrol, betulinic acid, and fluoxetine for a patient with recurrent, progressive anaplastic ependymoma.
Case Report
A 58-year-old male with recurrent spinal cord ependymoma (CNS WHO grade 3) presented with progressive leptomeningeal metastases and parenchymal cerebral metastases. Three and one-half years previously, an MRI of the spine disclosed an intrathecal mass measuring 2.2 x 1.4 x 3.5 cm mass located at L1-2 that was occupying the entire spinal canal and displacing and deforming the conus tip of the L. posterior margin of the thecal sac. L1-L2 laminectomy and resection of an intradural tumor were performed followed by craniospinal irradiation. Subsequently, the disease remained relatively stable until indolent progression 14 months after diagnosis led to the use of temozolomide-lapatinib. A new metastatic lesion measuring 8 mm lesion in Meckel’s cave was treated with gamma knife radiotherapy. The lesion was stabilized but not resolved. The patient was hospitalized for a nearly fatal Pneumocystis jirovecii pneumonia (PJP) precipitated by temozolomide use.
Eight months later, chemotherapy was held for progressive pancytopenia associated with a new diagnosis of T cell large granular leukemia (LGL), an indolent chronic leukemia. At 22 months from diagnosis, progressive disseminated ependymoma was noted in the thoracic spine at T8-9 along with multiple enhancing nodules involving the cauda equina and 4 new nodules in the occipital lobe. A persistent 6 mm lesion was noted in Meckel’s cave.
Genomic analysis
Next generation sequencing (NGS) was performed. (Tempus Inc.; Chicago, IL) Focal high-level amplification of the MYCN on chromosome 2p24 with 20 copies was observed, and a truncating frameshift mutation in the phosphatase and tensin homolog (PTEN) gene and loss-of-function mutation in ERCC excision repair 3 (ERCC3) nucleotide excision repair gene were identified. Chromosomal copy number analysis demonstrates gain of 14q, as well as losses of chromosomes 2p and 10. The combination of PTEN mutation and deletion implies biallelic loss of the tumor suppressor. No pathogenic mutations, focal amplifications, deep deletions, or structural variants were identified.
Course and treatment regimen
As mentioned, new metastatic lesions in the spine and brain were noted. Based on the signaling pathway impact analysis of MYCN and PTEN, the patient commenced the regimen listed in Table 2. Because, these naturopathic agents do not possess toxicity individually, they were combined without prior combinatorial trials to define safety and tolerability. The patient achieved disappearance of all lesions in supratentorial CNS, including the lesion in Meckel’s cave. The spinal lesions regressed substantially. No toxicity was observed from the treatment.
|
INTERVENTION |
TARGET |
SOURCE |
|---|---|---|
|
Tanshinone |
[miR-32, Let-7]↑ → AURKA↓ GPX4 |
Aqueous extract (i.e., tea) of danshen root, 76 ounces per day (~2 L), prepared each day from approximately 6 cm danshen root boiled in water on stove top, (purchased in China Town, Honolulu, HI)
|
|
Genistein |
PLK1↓ miR-34a↑ |
Genistein 1,000 mg three times per day, sourced from Fallon Pharmacy (Latham, NY)
|
|
Resveratrol |
AKT↓ AMPK↑ → MTOR↓ miR-21↓
|
Resveratrol capsules utilizing 1,000 mg po q8h |
|
Betulinic acid |
USP7↓ |
Chaga mushroom capsules were used as a source of betulinic acid
|
|
Fluoxetine
|
CSK1↓ |
Initiated at 20 mg daily and escalated by 20 mg increments in one-week intervals to 80 mg. |
At 6 months from inception, MRI identified a new left pre-pontine lesion. Craniotomy and sub-total resection through a retrosigmoid approach was performed, however the tumor was entangled with the 5th cranial nerve precluding complete resection. Pathology confirmed recurrent ependymoma, (CNS WHO grade 3). Repeat molecular profiling demonstrated a new chromosomal amplification of 11q22 with more than 10 copies of YES-associated protein-1(YAP1), baculoviral IAP repeat containing 2 (BIRC2), and BIRC3, representing a novel resistance mechanism for disease progression. Stereotactic body irradiation (SBRT) was administered to the remaining pre-pontine disease.
Discussion
In contrast to the strategies of attacking DNA with chemotherapy and radiation or blocking oncogenes with kinase inhibitors, synthetic lethal targeting capitalizes on “Achilles heel” vulnerabilities created by mutation or chromosomal copy number abnormalities, in this case MYCN amplification. Synthetic lethal partners define key nodes in a dysregulated protein network. As presented here, co-targeting three synthetic lethal partners in combination with takedown of PTEN-PI3K-AKT-MTOR and USP7 resistance pathways led to immediate, substantial disease regression for 6 months prior to the development of a new clone of disease sporting YAP1 and BIRC2/3 amplification to bypass the targeting described above.
Notably, the single agent use of the AURKA inhibitor alisertib failed in phase II trials to meet its prespecified endpoints in MYCN-driven neuroblastoma [49] and neuroendocrine prostate cancer [50].] However, the result is not surprising considering that N-Myc also upregulates PLK1 transcription [51]. The signaling pathway analysis depicts a redundant and self-amplifying disease network where both AURKA and PLK1 impact the cell cycle and inhibit FBXW7. This redundancy suggests co-targeting PLK1 and AURKA may be essential. AURKA phosphorylates PLK1 at Threonine-210 during late in G2 to promote checkpoint recovery and cause entry into prophase. In return, PLK1 promotes the recruitment of AURKA to the centrosomes in late G2 that is important for centrosome maturation and bipolar spindle formation during metaphase [52]. In addition to their mutual activation, PLK1 cooperates with AURKA to regulate other mitotic functions, including cohesion, centrosome maturation, and kinetochore–microtubule interactions [53]. PLK1 also has non-canonical targets beyond the cell cycle, including the MAP kinase pathway through RAF1 protooncogene, serine/threonine kinase (RAF1 -CRAF) phosphorylation [54], while AURKA promotes tumorigenesis by participating in epithelial-mesenchymal transition (EMT), metastasis, survival, and self-renewal of cancer stem cells [55]. Both kinases perpetrate signaling cascades that activate other transcription factors, including glioma-associated oncogene zinc finger protein (GLI1), hypoxia-inducible factor 1-alpha (HIF1A), zinc finger E-box-binding homeobox 1 (ZEB1), JUN, and FOXM1 by PLK1, and YAP1, nuclear factor kappa-light-chain-enhancer of activated B cells (NFKB), Forkhead box protein O1 (FOXO1), and signal transducer and activator of transcription 3 (STAT3) by AURKA to drive malignant behavior. Not surprisingly, laboratory models show that simultaneous inhibition of PLK1 and AURKA produces synergistic anti-tumor activity compared to targeting either kinase alone [56,57]. Based on these contemporary signaling pathway insights, co-targeting both PLK1 and AURKA targeting may be the minimum combinatorial strategy needed for approaching MYCN-driven cancers. By implication, the redundancies in MYCN signaling network suggests the principle of establishing single agent activity before advancing a drug for combination therapy may be molecularly naïve for diseases where simultaneous takedown of multiple nodes is a prerequisite for disabling redundant signaling.
With regard to N-Myc ubiquitin turnover, FBXW7 requires dual phosphorylation of N-Myc at T58 by GSK3-ß and at S62 by G2/mitotic-specific cyclin-B1 (CCNB1), both representing the CPD site [58,59]. However, upregulation of PI3K-AKT pathway associated with the bi-allelic loss of PTEN in this patient leads to unopposed PI3K activation of AKT signaling to cause inhibition of GSK3-ß and loss of T58 phosphorylation. The NCYM transcript from the antisense MYCN gene (MYCNOS) also directly inhibits GSK3-ß [60]. In parallel, GSK3-ß mediated phosphorylation is needed for the activation of another E3 ubiquitin kinase F-box/WD repeat-containing protein 1A (ß-TrCP - BTRC) [61]. Inhibition of ß-TrCP function accelerates movement past G2/M checkpoint and from metaphase to anaphase to precipitate mitotic slippage [62]. When ß-TrCP is compromised, its non-canonical targets, ß-catenin, YAP1, and DEP-domain containing mTOR-interacting protein (DEPTOR) (inhibitor of MTOR), also promote cancer progression [63], including activation of PP2A to dephosphorylate MYCN at S62, thus promoting its resistance to FBXW7. GSK3ß also phosphorylates the CPD of AURKA to promote its turnover by FBXW7.
The activation of AKT caused by PTEN loss in this cancer also inhibits tuberous sclerosis 1 and 2, hamartin and tuberin (TSC1/2) restraint of Ras homolog enriched in brain (RHEB)-mediated activation of MTOR and subsequently PP2A, a phosphatase which reverses S62 phosphorylation. RICTOR, also a target of FBXW7 [65], creates yet another feedforward loop by upregulating AKT to redouble GSK3-ß and FBXW7 inhibition [66]. RICTOR also triggers O-GlcNAc transferase or N-acetylglucosaminyltransferase (OGT) to cause O-GluNAcylation, a post-translational modification that activates numerous oncogenes, including PI3K, AKT, and YAP1 [67], thus further compounding the feed-forward inhibition of FBXW7. Consequently, PTEN loss in this patient’s disease resulting in inhibition of GSK3-ß causes broad dysregulation of numerous brakes on the cell cycle and the normal ubiquitin turnover of multiple oncogenes, including N-Myc. For this reason, AKT and MTOR represent critical nodes in this disease network. Because of PLK1 and AURKA also inhibit FBXW7 function, re-establishment of N-Myc ubiquitination depends upon the combination strategy with AKT and MTOR inhibition concurrently with PLK1 and AURKA blocking strategies. Though PLK1, AURKA, AKT, MTOR, FBXW7, and USP7 are genomically normal in this cancer, their integrated dysregulation and pleiotropic interactions in the proteome form a therapeutic imperative. Accordingly, the concomitant blockade strategy described here rescues FBXW7 ubiquitin turnover, re-establishes elimination of N-Myc by the proteasome, and precipitates cell cycle arrest, thereby achieving clinical efficacy by simultaneously overcoming key redundant resistance pathways and collapsing key driver mechanisms of the malignant phenotype (Figure 1).
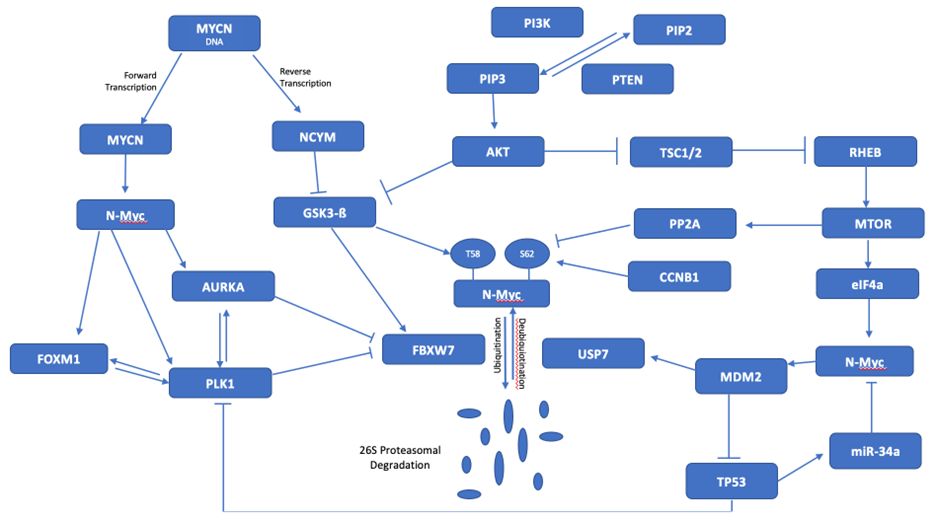
Figure 1. MYCN signaling network. The inhibition of FBXW7 by PLK1 and AURKA interacts with the dysregulated PTEN-AKT-GSK3-B and MTOR pathways to promote stabilization of N-Myc. MDM2 also generates stabilization of N-Myc by inhibiting TP53 and miR-34a.
MYCN-driven cancers also enhance cellular iron import [68]. The resulting increase in reactive oxygen species arising from the Fenton reaction threatens the cell with oxidative stress and requires glutathione peroxidase-4 (GPX4) to neutralize hydroxyl radicals. Normally, glutathione (GSH) mitigates oxidate stress and is consumed in the reaction to form oxidized glutathione (GSSG). However, GPX4 uses nicotinamide adenine dinucleotide phosphate (NADPH) to regenerate GSSG back to GSH, thus sustaining the antioxidant resistance by preventing the formation of lipid hydroperoxides that permeabilize cell membranes to cause ferroptosis (i.e., iron-dependent cell death, a non-apoptotic form of cell death). Hence, GPX4 is another SLP of MYCN.
Notably, tanshinone has pleiotropic antitumor effects. While conceived here as an AURKA inhibitor, it also functions as a GPX4 inhibitor [69]. Tanshinone can also enhance oxidative stress by impeding expression of cystine/glutamate transporter gene (SLC7A11) [70], the cystine/glutamate transporter protein (Xc) antiporter that mediates the rate limiting step in GSH biosynthesis, and by silencing the expression of Ferritin heavy chain (FTH1) [71], which lowers oxidative stress by sequestering iron. The resulting increase in oxidative stress caused by tanshinone has been shown to accentuate the effect of chemotherapy to cause ferroptosis, perhaps accounting for 50% of chemotherapy’s cytotoxic effect. Besides its effect on AURKA, a retrospective study looking at danshen use in patients with breast cancer in Taiwan showed it was associated with a marked improvement in survival (log-rank: p < 0.001) mediated by blocking GPX4 [72].
Notably, NRF2 (NFE2L2), the master regulator of antioxidant response element which prevents ferroptosis, is another FBXW7 ubiquitination target [73] rescued by the MYCN network. NRF2 also possesses a GSK3-ß CPD site [74,75]. Mechanistically, N-Myc upregulation of AURKA and PLK1 leads to oxidative stress resistance by inhibiting FBXW7 ubiquitination of NRF2. Conversely, GSK3-ß phosphorylation resulting from AKT inhibition prepares NRF2 for ubiquitin turnover, while rescue of FBXW7 from AURKA-mediated inhibition by tanshinone overcomes oxidative stress resistance by simultaneously enhancing NRF2 ubiquitination and GPX4 inhibition. Accordingly, tanshinone could have precipitated a lethal level of oxidative stress for this cancer, producing a cytotoxic effect to augment the collapse of the tumor network described above. As such, tanshinone could be developed as a component of a comprehensive strategy to eliminate oxidative stress resistance, including targeting GSH with sulfasalazine and thioredoxin blockade with auranofin to precipitate ferroptosis and overcome resistance to conventional therapeutic strategies [44]. Another redundant mechanism to preserve oxidative stress resistance involves GSK3-ß phosphorylation of NRF2 S338 which facilitates its ubiquitin turnover by ß-TrCp, again underscoring the importance of co-targeting AKT in the reversal of oxidative stress resistance [76]. Finally, FBXW7 rescue would enhance the proteasomal turnover of MCL1 and FOXM1, thereby reversing major chemotherapy and radiation resistance mechanisms arising from apoptotic blockade and enhanced DNA repair. For these reasons, upfront use of the network targeting in MYCN-amplified cancer described here could be employed to overcome multiple key resistance mechanisms to chemotherapy and radiation.
MYCN-driven cancers may be prone to chromosomal instability arising from replications stress caused by cell cycle dysregulation, compromise of G2/M checkpoint caused by PLK1 upregulation, and p53 takedown caused by upregulation of MDM2. The resulting chromosomal instability, i.e., genomic entropy, drives evolutionary drug resistance, mediated by copy number aberrations that create resistance pathways. In this case, the appearance of 11q22 amplification produces YAP1 whose dysregulation is tumorigenic and BIRC2/3, the inhibitor of apoptosis proteins (IAPs) that generate cell survival in the face of chemotherapy and radiation. Therefore, it is likely that the earliest possible use of this network targeting approach would improve its efficacy by beating the evolution of additional resistance mechanisms. MYCN-driven genomic instability makes the entropy clock tick faster, and eventually, a relentlessly evolving disease complexity and heterogeneity that is either undruggable or becomes insurmountable by drugs that reasonably could be combined. For this reason, the conventional requirement for efficacy in the salvage setting before considering a drug for upfront use represents another molecularly naïve criterion prone to eliminate potentially effective approaches that might have been effective in the setting of less complex early disease management.
The achievement of a 6-month disease remission with plant-derived substances is noteworthy and provides clinical validation of synthetic lethality observed in the laboratory. While ordinary cancer prevention supplement doses of plant-derived agents may not be adequate to achieve enzyme inhibition, many naturopathic agents possess attractive enzyme affinity constants. The large doses of naturopathic compounds used in this patient may be a likely factor in the clinical efficacy observed. The absence of toxicity is also noteworthy and may be due to the relatively inconsequential impact of these substances in normal tissues possessing intact PTEN and diploid MYCN.
The prior use of radiation and temozolomide and lapatinib had limited success for this patient, echoing the frustration of treating MYCN-driven cancers. Additionally, the patient’s compromised bone marrow function from T cell LGL was a relative contraindication to further chemotherapy. Practically, there are no clinical trials testing any PLK1, AURKA, AKT, and MTOR inhibitors in rare diseases like ependymoma, much less in the combination proposed here. The absence of a clinical trials option for this patient supported the naturopathic approach to meet the therapeutic imperatives determined by the sequencing results. But more importantly, drug development for MYCN-driven cancers has not caught up to the critical necessity of co-targeting multiple kinases in a complex disease network. To compound the challenge, no business model exists for co-developing a diverse group of pharmaceuticals simultaneously. But even if pharmaceuticals had been commercially available, the combination of multiple kinase inhibitors to target PLK1 (volasertib, rigosertib), AURKA (alisertib), AKT (ipatasertib), MTOR (everolimus, rapamycin), and USP7 (p5091) would have been a daunting or unfeasible proposition. Nevertheless, without embracing the complexity of the MYCN network and co-targeting redundancies and resistance mechanisms, single agents have no chance to achieve the desired goal. In this case, opening the therapeutic armamentarium to include plant-derived chemicals with a long track record of safety in other medical traditions permitted the design of a complex combinatorial regimen that achieves the goal of rationally co-targeting redundant disease drivers with minimal or no toxicity. By re-purposing naturopathic agents at doses suitable to achieve enzyme inhibition, the patient was able to bypass the shortcomings of drug development for the key drivers in MYCN-driven proteome. Potentially, the successful result presented here could re-energize the ambition of curing this notoriously lethal group of diseases and form a basis for a tumor site-independent, i.e., “basket,” clinical trial design of network-directed pharmaceutical-naturopathic combinations for MYCN-driven cancers. While we organize clinical trials to confirm the laboratory observations that led to the success in this case report, oncologists could consider this genomically-informed, apparently safe, and relatively inexpensive combination for MYCN disease management.
Abbreviations
AKT: Protein kinase B (PKB) gene; AMPK: 5' Adenosine Monophosphate-activated Protein Kinase; AURKA: Aurora Kinase A; BCL2: B-cell Lymphoma 2; BIRC2/3: Baculoviral IAP Repeat Containing 2/3; βTrCP: F-box/WD repeat-containing protein 1A; C-Myc: C-myc proto-oncogene protein; CCNB1: G2/mitotic-specific cyclin-B1; CCNE1/2: G1/S-specific cyclin-E1 and 2; CDC4: Cell Division Control protein 4; CDK2: Cyclin-Dependent Kinase 2; CDKN1: Cyclin-Dependent Kinase Inhibitor 1 gene; CDKN1B: Cyclin-Dependent Kinase Inhibitor 1B gene; CKS1: Casein Kinase-1; CPD: CDC4 Phosphodegron; DEPTOR: DEP-domain containing mTOR-interacting protein; ERCC3: ERCC excision repair 3; FBXW7: F-box/WD repeat-containing protein 7; FOXM1: Forkhead box protein M1; FOXO1: Forkhead box protein O1; FTH1: Ferritin Heavy Chain; GLI1: Glioma-associated oncogene zinc finger protein; GPX4: Glutathione Peroxidase 4; GSH: Glutathione; GSK3-ß: Glycogen Synthase Kinase beta; GSSG: Glutathione Disulfide; HIF1A: Hypoxia-Inducible Factor 1-alpha; JUN: Transcription factor Jun; KEAP1: Kelch-like ECH-Associated Protein 1; KLF5: Krueppel-Like factor 5; MCL1: Induced myeloid leukemia cell differentiation protein; MDM2: Mouse Double Minute 2 homolog E3 ubiquitin-protein ligase; miR-21: micro-RNA 21; MTOR: Mammalian Target Of Rapamicin; MYCN: N-myc proto-oncogene; N-Myc: N-myc proto-oncogene protein; NADPH: Nicotinamide Adenine Dinucleotide Phosphate; NFE2L2: Nuclear Factor Erythroid 2-related factor 2 gene; NFKB: Nuclear Factor Kappa-light-chain-enhancer of activated B cells; NOTCH1/2: Neurogenic locus Notch homolog protein 1 and 2; NRF2: Nuclear Factor erythroid 2-related Factor 2 protein; OGT: O-GlcNAc transferase or N-acetylglucosaminyltransferase; p21: Cyclin-dependent kinase inhibitor 1 protein; p27: Cyclin-dependent kinase inhibitor 1B protein; p53: Cellular tumor antigen p53; PARP: Poly ADP Ribose Polymerase; PDE4: Phosphodiesterase 4; PI3K: Phosphoinositide 3-Kinase; PLK1: Polo-Like Kinase; PTEN: Phosphatase and Tensin homolog; PP2A: Protein Phosphatase 2; RAF1: RAF1 protooncogene, serine/threonine kinase; RHEB: Ras Homolog Enriched in Brain; RICTOR: Rapamycin-Insensitive Companion of Mammalian Target of Rapamycin; SLC7A11: Cystine/glutamate transporter gene; SLP: Synthetic Lethal Partner; STAT3: Signal Transducer and Activator of Transcription 3; TSC1/2: Tuberous Sclerosis 1 and 2, hamartin and tuberin; USP7: Ubiquitin-Specific-processing Protease 7; Xc: Cystine/glutamate transporter protein; YAP1: YES-Associated Protein-1; ZEB1: Zinc finger E-box-binding homeobox 1
Conflict of Interest
I am the sole author of the manuscript and declare I have no conflicts of interest.
Funding Statement
This work received no funding support.
Consent
Permission was obtained from the patient to publish this case report.
References
2. Ghasemi DR, Sill M, Okonechnikov K, Korshunov A, Yip S, Schutz PW, et al. MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathologica. 2019 Dec;138(6):1075-89.
3. Raffeld M, Abdullaev Z, Pack SD, Xi L, Nagaraj S, Briceno N, et al. High level MYCN amplification and distinct methylation signature define an aggressive subtype of spinal cord ependymoma. Acta Neuropathologica Communications. 2020 Dec;8(1):1-1.
4. Otto T, Horn S, Brockmann M, Eilers U, Schüttrumpf L, Popov N, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell. 2009 Jan 6;15(1):67-78.
5. Liu X. Plk1, a negative regulator of p21?. Cell Cycle. 2009;8(3):335-337.
6. Song B, Liu XS, Davis K, Liu X. Plk1 phosphorylation of Orc2 promotes DNA replication under conditions of stress. Molecular and Cellular Biology. 2011 Dec 1;31(23):4844-56.
7. Zhang Z, Su WH, Feng C, Yu DH, Cui C, Xu XY, et al. Polo-like kinase 1 may regulate G2/M transition of mouse fertilized eggs by means of inhibiting the phosphorylation of Tyr15 of Cdc2. Molecular Reproduction and Development. 2007 Oct;74(10):1247-54.
8. Puig-Butille JA, Vinyals A, Ferreres JR, Aguilera P, Cabre E, Tell-Marti G, et al. AURKA overexpression is driven by FOXM1 and MAPK/ERK activation in melanoma cells harboring BRAF or NRAS mutations: impact on melanoma prognosis and therapy. Journal of Investigative Dermatology. 2017 Jun 1;137(6):1297-310.
9. Zhang Z, Zhang G, Kong C. FOXM1 participates in PLK1-regulated cell cycle progression in renal cell cancer cells. Oncology Letters. 2016 Apr 1;11(4):2685-91.
10. Fu Z, Malureanu L, Huang J, Wang W, Li H, Van Deursen JM, et al. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional Programme required for mitotic progression. Nature Cell Biology. 2008 Sep;10(9):1076-82.
11. Slack A, Chen Z, Tonelli R, Pule M, Hunt L, Pession A, et al. The p53 regulatory gene MDM2 is a direct transcriptional target of MYCN in neuroblastoma. Proceedings of the National Academy of Sciences. 2005 Jan 18;102(3):731-6.
12. He J, Gu L, Zhang H, Zhou M. Crosstalk between MYCN and MDM2-p53 signal pathways regulates tumor cell growth and apoptosis in neuroblastoma. Cell Cycle. 2011 Sep 1;10(17):2994-3002.
13. Navarro F, Lieberman J. miR-34 and p53: New Insights into a Complex Functional Relationship. PloS One. 2015 Jul 15;10(7):e0132767.
14. Tivnan A, Tracey L, Buckley PG, Alcock LC, Davidoff AM, Stallings RL. MicroRNA-34a is a potent tumor suppressor molecule in vivo in neuroblastoma. BMC Cancer. 2011 Dec;11(1):1-1.
15. Wei JS, Song YK, Durinck S, Chen QR, Cheuk AT, Tsang P, et al. The MYCN oncogene is a direct target of miR-34a. Oncogene. 2008 Sep;27(39):5204-13.
16. Wang X, Sun Q. TP53 mutations, expression and interaction networks in human cancers. Oncotarget. 2017 Jan 3;8(1):624.
17. Wang X, Simon R. Identification of potential synthetic lethal genes to p53 using a computational biology approach. BMC Medical Genomics. 2013 Dec;6(1):1-0.
18. Ando K, Ozaki T, Yamamoto H, Furuya K, Hosoda M, Hayashi S, et al. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. Journal of Biological Chemistry. 2004 Jun 11;279(24):25549-61..
19. Gu L, Zhang H, He J, Li J, Huang M, Zhou M. MDM2 regulates MYCN mRNA stabilization and translation in Human Neuroblastoma Cells. Oncogene. 2012 Mar;31(11):1342-53.
20. He J, Gu L, Zhang H, Zhou M. Crosstalk between MYCN and MDM2-p53 signal pathways regulates tumor cell growth and apoptosis in Neuroblastoma. Cell Cycle. 2011 Sep 1;10(17):2994-3002.
21. Tavana O, Li D, Dai C, Lopez G, Banerjee D, Kon N, et al. HAUSP deubiquitinates and stabilizes N-Myc in neuroblastoma. Nature Medicine. 2016 Oct;22(10):1180-6.
22. Wang D, Pierce A, Veo B, Fosmire S, Danis E, Donson A, et al. A regulatory loop of FBXW7-MYC-PLK1 controls tumorigenesis of MYC-driven medulloblastoma. Cancers. 2021 Jan 21;13(3):387.
23. Liu Z, Chen SS, Clarke S, Veschi V, Thiele CJ. Targeting MYCN in pediatric and adult cancers. Frontiers in Oncology. 2021 Feb 8;10:623679.
24. Yeh CH, Bellon M, Nicot C. FBXW7: a critical tumor suppressor of human cancers. Molecular Cancer. 2018 Dec;17(1):1-9.
25. Chen Y, Li Y, Xue J, Gong A, Yu G, Zhou A, et al. Wnt‐induced deubiquitination FoxM1 ensures nucleus β‐catenin transactivation. The EMBO journal. 2016 Mar 15;35(6):668-84.
26. Liao GB, Li X-Z, Zeng S, et al. Regulation of the master regulator FOXM1 in cancer. Cell Commun Signal. 2018;16(1):57.
27. Zona S, Bella L, Burton MJ. Nestal de Moraes G, Lam EW. FOXM1: an emerging master regulator of DNA damage response and genotoxic agent resistance. Biochim Biophys Acta. 2014 Nov;1839(11):1316-22.
28. Shang X, Burlingame SM, Okcu MF, Ge N, Russell HV, Egler RA, et al. Aurora A is a negative prognostic factor and a new therapeutic target in human neuroblastoma. Molecular Cancer Therapeutics. 2009 Aug;8(8):2461-9.
29. O'Neil NJ, Bailey ML, Hieter P. Synthetic lethality and cancer. Nature Reviews Genetics. 2017 Oct;18(10):613-23.
30. Bi J, Khan A, Tang J, Armando AM, Wu S, Zhang W, et al. Targeting glioblastoma signaling and metabolism with a re-purposed brain-penetrant drug. Cell Reports. 2021 Nov 2;37(5):109957.
31. Thng DK, Toh TB, Chow EK. Capitalizing on synthetic lethality of MYC to treat cancer in the digital age. Trends in Pharmacological Sciences. 2021 Mar 1;42(3):166-82..
32. Bogen D, Wei JS, Azorsa DO, Ormanoglu P, Buehler E, Guha R, et al. Aurora B kinase is a potent and selective target in MYCN-driven neuroblastoma. Oncotarget. 2015 Nov 11;6(34):35247.
33. King D, Li XD, Almeida GS, Kwok C, Gravells P, Harrison D, et al. MYCN expression induces replication stress and sensitivity to PARP inhibition in Neuroblastoma. Oncotarget. 2020 Jun 6;11(23):2141..
34. Mathew S, Vazhappilly CG. Recent pharmacological advances on genistein in clinical trials. EXCLI journal. 2020 Aug 5;19:1120-3.
35. Shin SB, Woo SU, Chin YW, Jang YJ, Yim H. Sensitivity of TP53-mutated cancer cells to the phytoestrogen genistein is associated with direct inhibition of Plk1 activity. journal of Cellular Physiology. 2017 Oct;232(10):2818-28.
36. Ullmann U, Metzner J, Frank T, Cohn W and Riegger C. Safety, tolerability, and pharmacokinetics of single ascending doses of synthetic genistein (Bonistein) in healthy volunteers. Adv Ther. 2005 Jan-Feb;22(1):65-78.
37. Wei JS, Song YK, Durinck S, Chen QR, Cheuk AT, Tsang P, et al. The MYCN oncogene is a direct target of miR-34a. Oncogene. 2008 Sep;27(39):5204-13.
38. Xia J, Duan Q, Ahmad A, Bao B, Banerjee S, Shi Y, et al. Genistein inhibits cell growth and induces apoptosis through up-regulation of miR-34a in pancreatic cancer cells. Curr Drug Targets. 2012 Dec 1;13(14):1750-6.
39. Ma ZL, Zhang BJ, Wang DT, Li X, Wei JL, Zhao BT, et al. Tanshinones suppress AURKA through up-regulation of miR-32 expression in non-small cell lung cancer. Oncotarget. 2015 Aug 21;6(24):20111.
40. Ma ZL, Zhang BJ, Wang DT, Li X, Wei JL, Zhao BT, et al. Tanshinones suppress AURKA through up-regulation of miR-32 expression in non-small cell lung cancer. Oncotarget. 2015 Aug 21;6(24):20111.
41. Liu X, Zou H, Zhao Y, Chen H, Liu T, Wu Z, et al. Tanshinone Inhibits NSCLC by Downregulating AURKA Through Let-7a-5p. Front Genet. 2020 Aug 7;11:838.
42. Krishnan A, Hariharan R, Nair SA and Pillai MR. Fluoxetine mediates G0/G1 arrest by inducing functional inhibition of cyclin dependent kinase subunit (CKS)1. Biochem Pharmacol. 2008 May 15;75(10):1924-34.
43. Park SJ, Ahmad F, Philp A, Baar K, Williams T, Luo H, et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell. 2012 Feb 3;148(3):421-33.
44. Roy SK, Chen Q, Fu J, Shankar S, Srivastava RK. Resveratrol inhibits growth of orthotopic pancreatic tumors through activation of FOXO transcription factors. PLoS ONE. 2011 Sep 27;6(9):e25166.
45. Liu P, Liang H, Xia Q, Li P, Kong H, Lei P, et al. Resveratrol induces apoptosis of pancreatic cancers cells by inhibiting miR-21 regulation of BCL-2 expression. Clin Transl Oncol. 2013 Sep;15(9):741-6.
46. Grunblatt E, Wu N, Zhang H, Liu X, Norton JP, Ohol Y, et al. MYCN drives chemoresistance in small cell lung cancer while USP7 inhibition can restore chemosensitivity. Genes Dev. 2020 Sep 1;34(17-18):1210-26.
47. Reiner T, Parrondo R, de Las Pozas A, Palenzuela D and Perez-Stable C. Betulinic acid selectively increases protein degradation and enhances prostate cancer-specific apoptosis: possible role for inhibition of deubiquitinase activity. PLoS One. 2013 Feb 12;8(2):e56234.
48. Jing B, Liu M, Yang L, Cai HY, Chen JB, Li ZX, et al. Characterization of naturally occurring pentacyclic triterpenes as novel inhibitors of deubiquitinating protease USP7 with anticancer activity in vitro. Acta Pharmacologica Sinica. 2018 Mar;39(3):492-8.
49. Mossé YP, Fox E, Teachey DT, Reid JM, Safgren SL, Carol H, et al. A Phase II Study of Alisertib in Children with Recurrent/Refractory Solid Tumors or Leukemia: Children's Oncology Group Phase I and Pilot Consortium (ADVL0921). Clin Cancer Res. 2019 Jun 1;25(11):3229-38.
50. Beltran H, Oromendia C, Danila DC, Montgomery B, Hoimes C, Szmulewitz RZ, et al. A Phase II Trial of the Aurora Kinase A Inhibitor Alisertib for Patients with Castration-resistant and Neuroendocrine Prostate Cancer: Efficacy and Biomarkers. Clin Cancer Res. 2019 Jan 1;25(1):43-51.
51. Xiao D, Yue M, Su H, Ren P, Jiang J, Li F, et al. Polo-like kinase-1 regulates Myc stabilization and activates a feedforward circuit promoting tumor cell survival. Molecular Cell. 2016 Nov 3;64(3):493-506..
52. Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, et.al. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008 Sep;455(7209):119-23.
53. Lens SM, Voest EE, Medema RH. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev Cancer. 2010 Dec;10(12):825-41.
54. Jianguo Wu, Andrei I Ivanov, Paul B Fisher, Zheng Fu. (2016) Polo-like kinase 1 induces epithelial-to-mesenchymal transition and promotes epithelial cell motility by activating CRAF/ERK signaling. 2016 Mar 22;5: e10734.
55. Du, R., Huang, C., Liu, K. et al. Targeting AURKA in Cancer: molecular mechanisms and opportunities for Cancer therapy. Mol Cancer. 2021 Dec;20(1):1-27.
56. Li J, Hong MJ, Chow JP, Man WY, Mak JP, Ma HT, et.al. Co-inhibition of polo-like kinase 1 and Aurora kinases promotes mitotic catastrophe. Oncotarget. 2015 Apr 4;6(11):9327.
57. Metselaar DS, du Chatinier A, Meel MH, et al., AURKA and PLK1 inhibition selectively and synergistically block cell cycle progression in diffuse midline glioma. iScience, 2022 May 13:104398.
58. Liu R, Shi P, Wang Z, Yuan C, Cui H. Molecular MYCN Dysregulation in Cancers. Front Oncol. 2021 Feb 3;10:625332.
59. Richards MW, Burgess SG, Poon E, Carstensen A, Eilers M, Chesler L, et al. Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proc Natl Acad Sci 2016 Nov 29;113(48):13726-31.
60. Suenaga Y, Islam SMR, Alagu J, Kaneko Y, Kato M, Tanaka Y , et al. NCYM, a cis-antisense gene of MYCN, encodes a de novo evolved protein that inhibits GSK3β resulting in the stabilization of MYCN in human neuroblastomas. PloS Genet 2014 Jan 2;10(1): e1003996.
61. Gao C, Chen G, Romero G, Moschos S, Xu X, Hu J. Induction of Gsk3β-β-TrCP interaction is required for late phase stabilization of β-catenin in canonical Wnt signaling. J Biol Chem. 2014 Mar 7;289(10):7099-108.
62. Lindqvist A, Rodríguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol 2009 Apr 20;185(2):193-202.
63. Wang Q, Zhou Y, Rychahou P, Harris JW, Zaytseva YY, Liu J, Wang C, et.al. Deptor Is a Novel Target of Wnt/β-Catenin/c-Myc and Contributes to Colorectal Cancer Cell Growth. Cancer Res. 2018 Jun 15;78(12):3163-75.
64. Kwon YW, Kim IJ, Wu D, Lu J, Stock WA, Liu Y, Huang Y, et.al. Regulates Aurora-A and Cooperates with Fbxw7 in Modulating Radiation-Induced Tumor Development. Mol Cancer Res 2012 Jun 1;10(6):834-44.
65. Koo J, Wu X, Mao Z, Khuri FR and Sun SY. Rictor Undergoes Glycogen Synthase Kinase 3 (GSK3)-dependent, FBXW7-mediated Ubiquitination and Proteasomal Degradation. J Biol Chem. 2015 May 29;290(22):14120-9.
66. Laugier F, Finet-Benyair A, André J, Rachakonda PS, Kumar R, Bensussan A, et.al. RICTOR involvement in the PI3K/AKT pathway regulation in melanocytes and melanoma. Oncotarget. 2015 Sep 9;6(29):28120.
67. Very N, Vercoutter-Edouart AS, Lefebvre T, Hardivillé S, El Yazidi-Belkoura I. Cross-Dysregulation of O-GlcNAcylation and PI3K/AKT/mTOR Axis in Human Chronic Diseases. Front Endocrinol (Lausanne). 2018 Oct 9;9:602.
68. Floros KV, Cai J, Jacob S, Kurupi R, Fairchild CK, Shende M, et.al. MYCN-Amplified Neuroblastoma Is Addicted to Iron and Vulnerable to Inhibition of the System Xc-/Glutathione Axis. Cancer Res. 2021 Apr 1;81(7):1896-908.
69. Wu CY, Yang YH, Lin YS, Chang GH, Tsai MS, Hsu CM, et.al. Dihydroisotanshinone I induced ferroptosis and apoptosis of lung cancer cells. Biomed Pharmacother. 2021 Jul 1; 139:111585.
70. Guan Z, Chen J, Li X and Dong N. Tanshinone IIA induces ferroptosis in gastric cancer cells through p53-mediated SLC7A11 down-regulation. Biosci Rep. 2020 Aug 28;40(8).
71. Mao W, Ding J, Li Y, Huang R and Wang B. Inhibition of cell survival and invasion by Tanshinone IIA via FTH1: A key therapeutic target and biomarker in head and neck squamous cell carcinoma. Exp Ther Med 2022 Aug 1;24(2):1-2.
72. Lin YS, Shen YC, Wu CY, Tsai YY, Yang YH, Lin YY et al. Danshen Improves Survival of Patients With Breast Cancer and Dihydroisotanshinone I Induces Ferroptosis and Apoptosis of Breast Cancer Cells . Front Pharmacol, 2019 Oct 31;10:1226.
73. Kar R, Jha SK, Ojha S, Sharma A, Dholpuria S, Raju VS, et al. The FBXW7-NOTCH interactome: A ubiquitin proteasomal system-induced crosstalk modulating oncogenic transformation in human tissues. Cancer Rep (Hoboken). 2021 Aug;4(4):e1369.
74. Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD et al. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene. 2013 Aug; 32(32):3765-81.
75. Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011; 31:1121-1133.
76. Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013 Aug;32(32):3765-81.
77. Gheghiani L, Loew D, Lombard B, Mansfeld J and Gavet O. PLK1 Activation in Late G2 Sets Up Commitment to Mitosis. 2017, Cell Reports 2017 Jun 6;19(10): 2060-73.
78. Liu X. Plk1, a negative regulator of p21? Cell Cycle. 2009;8(3):335-337.
79. Den Hollander J, Rimpi S, Doherty JR, Rudelius M, Buck A and Hoellein A. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood 2010 Sep 2;116(9):1498-505.
80. Toyoshima-Morimoto F, Taniguchi E and Nishida E. Plk1 promotes nuclear translocation of human Cdc25C during prophase. EMBO Rep. 2002 Apr;3(4):341-8.
81. García-Gutiérrez L, Bretones G, Molina E, Arechaga I, Symonds C, Acosta JC, et al. Myc stimulates cell cycle progression through the activation of Cdk1 and phosphorylation of p27. Sci Rep. 2019 Dec 10;9(1):1-7.
82. Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, et al. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001 Oct 5;294(5540):173-7.
83. O'Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007 Aug 6;204(8):1813-24.
84. Zhao D, Zheng HQ, Zhou Z, Chen C. The Fbw7 tumor suppressor targets KLF5 for ubiquitin-mediated degradation and suppresses breast cell proliferation. Cancer Res. 2010 Jun 1;70(11):4728-38.
85. Luan Y and Wang P. FBW7-mediated ubiquitination and degradation of KLF5. World J Biol Chem. 2014 May 5;5(2):216.
86. Koo J, Wu X, Mao Z, Khuri FR and Sun SY. Rictor Undergoes Glycogen Synthase Kinase 3 (GSK3)-dependent, FBXW7-mediated Ubiquitination and Proteasomal Degradation. J Biol Chem. 2015 May 29;290(22):14120-9.
87. Nateri AS, Riera-Sans L, Da Costa C, Behrens A. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science. 2004 Feb 27;303(5662):1374-8.
88. Li J, Pauley AM, Myers RL, Shuang R, Brashler JR, Yan R. SEL-10 interacts with presenilin 1, facilitates its ubiquitination, and alters A-beta peptide production. J Neurochem. 2002 Sep;82(6):1540-8.
89. McKenzie L, King S, Marcar L, Nicol S, Dias SS, Schumm K, et al. p53-dependent repression of polo-like kinase-1 (PLK1). Cell Cycle. 2010 Oct 15;9(20):4200-12.
90. Ito Y, Inoue A, Seers T, Hato Y, Igarashi A, Toyama T, et al. Identification of targets of tumor suppressor microRNA-34a using a reporter library system. Proc Natl Acad Sci U S A. 2017 Apr 11;114(15):3927-32.
91. Zhang L, Liao Y, Tang L. MicroRNA-34 family: a potential tumor suppressor and therapeutic candidate in cancer. J Exp Clin Cancer Res 2019 Dec;38(1):1-3.
92. Liu C, Kato Y, Zhang Z, Do VM, Yankner BA, He X. beta-Trcp couples beta-catenin phosphorylation-degradation and regulates Xenopus axis formation. Proc Natl Acad Sci U S A. 1999 May 25;96(11):6273-8.
93. Gao D, Inuzuka H, Tan MK, Fukushima H, Locasale JW, Liu P, et al. mTOR Drives Its Own Activation via SCFbTrCp-Dependent Degradation of the mTOR Inhibitor DEPTOR. Molecular Cell. 2011 Oct 21;44(2):290-303.
94. Catena V and Fanciulli M. Deptor: not only a mTOR inhibitor. J Exp Clin Cancer Res. 2017;36(1):12. Published 2017 Dec;36(1):1-9.
95. Biswas M, Phan D, Watanabe M and Chan JY. The Fbw7 tumor suppressor regulates nuclear factor E2-related factor 1 transcription factor turnover through proteasome-mediated proteolysis. J Biol Chem. 2011 Nov 11;286(45):39282-9.
96. Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011 Mar 15;31(6):1121-33.
97. Hsieh CH, Hsieh HC, Shih FS, Wang PW, Yang LX, Shieh DB, et al. An innovative NRF2 nano-modulator induces lung cancer ferroptosis and elicits an immunostimulatory tumor microenvironment. Theranostics. 2021;11(14):7072.
98. Choi M, Kim W, Cheon MG, Lee CW and Kim JE. Polo-like kinase 1 inhibitor BI2536 causes mitotic catastrophe following activation of the spindle assembly checkpoint in non-small cell lung cancer cells. Cancer Lett. 2015 Feb 28;357(2):591-601.
99. Gheghiani L, Wang L, Zhang Y, Moore XT, Zhang J, Smith SC, et al. PLK1 Induces Chromosomal Instability and Overrides Cell-Cycle Checkpoints to Drive Tumorigenesis. Cancer Res. 2021 Mar 1;81(5):1293-307.
100. Chen Y, Li Y, Xue J, Gong A, Yu G, Zhou A, et al. Wnt-induced deubiquitination FoxM1 ensures nucleus beta-catenin transactivation. EMBO J. 2016 Mar 15;35(6):668-84.
101. Fu Z, Malureanu L, Huang J, Wang W, Li H, van Deursen JM, et al. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat Cell Biol. 2008;10:1076-82.
102. Liao GB, Li XZ, Zeng S, Liu C, Yang SM, Yang L, et al. Regulation of the master regulator FOXM1 in cancer. Cell Communication and Signaling. 2018 Dec;16(1):1-5.
103. Zhang W, Fletcher L, Muschel RJ. The role of Polo-like kinase 1 in the inhibition of centrosome separation after ionizing radiation. Journal of Biological Chemistry. 2005 Dec 30;280(52):42994-9.
104. Dai Y, Liu S, Zhang WQ, Yang YL, Hang P, Wang H, et al. YAP1 regulates ABCG2 and cancer cell side population in Human Lung Cancer Cells. Oncotarget. 2017 Jan 17;8(3):4096.
105. Gujral TS, Kirschner MW. Hippo pathway mediates resistance to cytotoxic drugs. Proceedings of the National Academy of Sciences. 2017 May 2;114(18):E3729-38.
106. Close V, Close W, Kugler SJ, Reichenzeller M, Yosifov DY, Bloehdorn J, et al. FBXW7 mutations reduce binding of NOTCH1, leading to cleaved NOTCH1 accumulation and target gene activation in CLL. Blood, The Journal of the American Society of Hematology. 2019 Feb 21;133(8):830-9.
107. Jenkinson S, Koo K, Mansour MR, Goulden N, Vora A, Mitchell C, et al. Impact of NOTCH1/FBXW7 mutations on outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on the MRC UKALL 2003 trial. Leukemia. 2013 Jan;27(1):41-7.
108. Xiao D, Yue M, Su H, Ren P, Jiang J, Li F, et al. Polo-like kinase-1 regulates Myc stabilization and activates a feedforward circuit promoting tumor cell survival. Molecular Cell. 2016 Nov 3;64(3):493-506.
109. Ren Y, Bi C, Zhao X, Lwin T, Wang C, Yuan J, et al. PLK1 stabilizes a MYC-dependent kinase network in aggressive B cell lymphomas. The JOURNAL of Clinical Investigation. 2018 Dec 3;128(12):5517-30.
110. Otto T, Horn S, Brockmann M, Eilers U, Schüttrumpf L, Popov N, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell. 2009 Jan 6;15(1):67-78.
111. Wang D, Pierce A, Veo B, Fosmire S, Danis E, Donson A, et al. A regulatory loop of FBXW7-MYC-PLK1 controls tumorigenesis of MYC-driven medulloblastoma. CANCERS. 2021 Jan 21;13(3):387.
112. Liu Z, Chen SS, Clarke S, Veschi V, Thiele CJ. Targeting MYCN in pediatric and adult cancers. Frontiers in Oncology. 2021 Feb 8;10:623679.
113. Liu R, Shi P, Wang Z, Yuan C, Cui H. Molecular mechanisms of MYCN dysregulation in cancers. Frontiers in Oncology. 2021 Feb 3;10:625332.
114. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005 Feb 18;307(5712):1098-101.
115. Laugier F, Finet-Benyair A, André J, et al. RICTOR involvement in the PI3K/AKT pathway regulation in melanocytes and melanoma. Oncotarget. 2015;6(29):28120-28131.
116. Puig-Butille JA, Vinyals A, Ferreres JR, Aguilera P, Cabre E, Tell-Marti G, et al. AURKA overexpression is driven by FOXM1 and MAPK/ERK activation in melanoma cells harboring BRAF or NRAS mutations: impact on melanoma prognosis and therapy. Journal of Investigative Dermatology. 2017 Jun 1;137(6):1297-310..
117. Hong CS, Ho W, Zhang C, Yang C, Elder JB, Zhuang Z. LB100, a small molecule inhibitor of PP2A with potent chemo-and radio-sensitizing potential. Cancer Biology & Therapy. 2015 Jun 3;16(6):821-33.
118. Janghorban M, Farrell AS, Allen-Petersen BL, Pelz C, Daniel CJ, Oddo J, et al. Targeting c-MYC by antagonizing PP2A inhibitors in breast cancer. Proceedings of the National Academy of Sciences. 2014 Jun 24;111(25):9157-62..
119. Vaughan L, Clarke PA, Barker K, Chanthery Y, Gustafson CW, Tucker E, et al. Inhibition of mTOR-kinase destabilizes MYCN and is a potential therapy for MYCN-dependent tumors. Oncotarget. 2016 Sep 9;7(36):57525.
120. Kling MJ, Griggs CN, McIntyre EM, Alexander G, Ray S, Challagundla KB, et al. Synergistic efficacy of inhibiting MYCN and mTOR signaling against neuroblastoma. BMC Cancer. 2021 Dec;21(1):1-3.
121. Slack A, Lozano G, Shohet JM. MDM2 as MYCN transcriptional target: implications for neuroblastoma pathogenesis. Cancer letters. 2005 Oct 18;228(1-2):21-7.
122. He J, Gu L, Zhang H, Zhou M. Crosstalk between MYCN and MDM2-p53 signal pathways regulates tumor cell growth and apoptosis in neuroblastoma. Cell Cycle. 2011 Sep 1;10(17):2994-3002..
123. Tavana O, Li D, Dai C, Lopez G, Banerjee D, Kon N, et al. HAUSP deubiquitinates and stabilizes N-Myc in neuroblastoma. Nature Medicine. 2016 Oct;22(10):1180-6.
124. Wei JS, Song YK, Durinck S, Chen QR, Cheuk AT, Tsang P, et al. The MYCN oncogene is a direct target of miR-34a. Oncogene. 2008 Sep;27(39):5204-13.
125. Thor T, Künkele A, Pajtler KW, Wefers AK, Stephan H, Mestdagh P, et al. M i R-34a deficiency accelerates medulloblastoma formation in vivo. International Journal of Cancer. 2015 May 15;136(10):2293-303.
126. Zhang L, Liao Y, Tang L. MicroRNA-34 family: a potential tumor suppressor and therapeutic candidate in cancer. Journal of Experimental & Clinical Cancer Research. 2019 Dec;38(1):1-3.
127. Ferrarelli LK. Targeting AURKA to drug the undruggable. Science Signaling. 2016 Aug 2;9(439):ec175-
128. Minegishi M, Tachibana K, Sato T, Iwata S, Nojima Y, Morimoto C. Structure and function of Cas-L, a 105-kD Crk-associated substrate-related protein that is involved in beta 1 integrin-mediated signaling in lymphocytes. The Journal of Experimental Medicine. 1996 Oct 1;184(4):1365-75..
129. Pugacheva EN, Golemis EA. The focal adhesion scaffolding protein HEF1 regulates activation of the Aurora-A and Nek2 kinases at the centrosome. Nature Cell Biology. 2005 Oct;7(10):937-46.
130. Dias SS, Hogan C, Ochocka AM, Meek DW. Polo-like kinase-1 phosphorylates MDM2 at Ser260 and stimulates MDM2-mediated p53 turnover. FEBS letters. 2009 Nov 19;583(22):3543-8.
131. Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, et al. SCFFBW7 regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011 Mar;471(7336):104-9.
132. He J, Gu L, Zhang H, Zhou M. Crosstalk between MYCN and MDM2-p53 signal pathways regulates tumor cell growth and apoptosis in neuroblastoma. Cell Cycle. 2011 Sep 1;10(17):2994-3002.
133. Louwen F, Yuan J. Battle of the eternal rivals: restoring functional p53 and inhibiting Polo-like kinase 1 as cancer therapy. Oncotarget. 2013 Jul 13;4(7):958-71.
134. Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCFβ-TRCP. GENES & Development. 2010 Jan 1;24(1):72-85.
135. Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R, et al. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008 Sep 12;321(5895):1499-502.
136. Fan J, Bellon M, Ju M, Zhao L, Wei M, Fu L, et al. Clinical significance of FBXW7 loss of function in human cancers. Molecular CANCER. 2022 Dec;21(1):1-26.