Abstract
Ovarian cancer stands as the most lethal gynecologic malignancy and remains the fifth most common gynecologic cancer. Poor prognosis and low five-year survival rate are attributed to nonspecific symptoms at early phases along with a lack of effective treatment at advanced stages. It is thus paramount, that ovarian carcinoma be viewed through several lenses in order to gain a thorough comprehension of its molecular pathogenesis, epidemiology, histological subtypes, hereditary factors, diagnostic approaches, and methods of treatment. Above all, it is crucial to dissect the role that the unique peritoneal tumor microenvironment plays in ovarian cancer progression and metastasis. This short communication seeks to underscore several important aspects of the PI3K/AKT/mTOR/NFκB pathway in the context of ovarian cancer and discuss recent advances in targeting this pathway.
Keywords
PI3K/AKT/mTOR/NFκB, Ovarian cancer, Therapeutic implication
Phosphoinositol 3 Kinase (PI3K)
Phosphoinositol 3 kinase (PI3K) defines a class of lipid kinases that have the ability to phosphorylate the inositol ring 3′-OH group in inositol phospholipids and therefore produce phosphatidylinositol (3,4,5)-trisphosphate (PIP3) [1]. PI3K encompasses a family of enzymes divided into: Class IA PI3K which includes three isomers (α, β, δ) and Class IB which include the group (γ) [2,3]. PI3K Class IA is comprised of a regulatory subunit p85 along with a catalytic p110α subunit [4]. Mutations in the gene encoding the catalytic subunit of PI3K p110α, PIK3CA, are found in nearly 33% of clear-cell carcinoma cases [5,6], 20% of endometrioid and clear-cell carcinomas [7], 18–28% of cases of serous cystadenocarcinoma, with enhancement of the signature of activated PI3K in the majority of ovarian cancer cases irrespective of the subtype [8]. PIK3CA mutations are considered driver mutations that provide transformative advantages for high grade serous cancer (HGSC) [9]. Multivariate survival analysis revealed that PI3K protein expression was associated with poor survival in advanced HGSC [10]. In addition, several studies have shown that the rate of mutations in the PI3K pathway, especially in AKT and p70S6K, including missense mutations and amplifications, is correlated with higher rates of chemoresistance [11,12]. Chemo-sensitization could be achieved via downregulation of PI3K and/or its downstream effectors, AKT and mTORC1 [13-15]. The increased activation of PI3K in OvCa and its role as a hub for several cancerpromoting pathways, explain its many implications in cancer progression including oncogenic transformation, cell proliferation, adhesion, and apoptosis, as well as multiple metabolic pathways, thus aptly positioning it as a target for therapeutic advancement [16-19].
Protein Kinase B PKB/AKT
The AKT/PKB family comprises a group of serine threonine kinases, which are cAMP- and cGMP-dependent [20]. Three AKT isoforms have been identified: AKT1 (PKBα), AKT2 (PKBβ), and AKT3 (PKBγ) [1,21]. AKT1 is involved in cellular growth, angiogenesis, and tumor cell invasiveness. AKT is the main kinase which integrates upstream signals from PI3K and mammalian target of rapamycin complex 2 (mTORC2) with downstream signals to mTORC1 with subsequent activation of downstream substrates that induce cell cycle progression, protein synthesis, and cell growth [21], and dictate several cellular activities such as survival, proliferation, and migration [18,20,22]. Moreover, AKT promotes protein synthesis and cell growth through inhibition of tuberous sclerosis complex 2 (TSC2), and 4E-binding protein 1 (4E-BP1), that inhibit cell growth in various cancer types, and regulate mRNA translation and cellular proliferation, respectively [17,21,23-25]. AKT is inhibited by tumor suppressors including phosphatase and tensin homolog (PTEN) and inositol polyphosphate 4-phosphatase type II (INPP4B). In ovarian cancer, AKT1 is mutated and AKT2 is amplified in about 40% [17,26]. Overexpression of AKT in OvCa is associated with advanced stageplatinum resistance [12,27]. Furthermore, data curated from The Cancer Genome Atlas (TCGA) revealed that the expression of AKT1, AKT2, and AKT3 was associated with poor patient survival [28].
Mammalian Target of Rapamycin (mTOR)
mTOR comprises two biochemically and functionally independent catalytic complexes, mTORC1 and mTORC2. Both mTOR complexes are implicated in the induction of angiogenesis, proliferation, and cellular survival [2,29]. Phospho-mTOR activates two downstream targets: 4E-binding protein 1 (4E-BP1) and ribosomal protein S6 kinase (S6K). In aggressive cancers, 4E-BP1 functions as a hypoxia-inducible switch, allowing for translation of factors, and hence facilitating angiogenesis and antiapoptotic cell growth [25,30]. Phosphorylated S6K is required for cell growth and G1 cell cycle progression [31,32]. mTORC1 is activated and overexpressed along with its downstream effectors, 4EBP1 and p70S6K, in advanced HGSC [8,33] warranting the use of mTOR inhibitors as targeted therapies in several clinical trials [34-37]. Consistently, analysis of TCGA data indicated that high expression of mTOR is associated with poor survival rates in patients with advanced stage HGSC.
Nuclear factor-κ light chain enhancer of activated B cells (NFκB)
Nuclear factor-κ light chain enhancer of activated B cells (NFκB) encompasses a group of transcription factors that are divided into two classes: Class I, which include NFκB1 or p50/p105, and NFκB2 or p52/p100. Class II includes RelA/p65, RelB, and c-Rel [38,39]. The NFκB canonical pathway includes NFκB, IkBα, and RelA/p65. IkBα is phosphorylated by the Inhibitor of Nuclear Factor Kappa B Kinase (IKK) [40,41]. Upon phosphorylation of NFκB1, the IkBα subunit undergoes ubiquitination and subsequent proteasomal degradation. This allows p50 and p65/RelA to dimerize and translocate to the nucleus where the heterodimer induces transcription of genes involved in inflammation, cell growth, chemoresistance, and apoptosis [42-44]. Alternatively, the non-canonical pathway is activated when inflammatory cytokines, TNF and IL1, bind to their respective receptors and subsequently signal to NFκB inducing kinase (NIK) to activate the IKK complex [45]. High expression of the p65/RelA subunit of NFκB, along with cleaved caspase 3 confers poor outcomes in OvCa patients [46]. Jinawath et al., [44] demonstrated that inhibition of NFκB resulted in enhanced efficacy of cisplatin in vitro and in vivo OvCa models. Upregulation of the p65/RelA subunit of NFκB increased the resistance of OvCa to carboplatin [44], and significantly enhanced the aggressiveness of OvCa cells [47]. Our earlier report [28] showed that in HGSC data from TCGA, the expression of NFκB subunits, p65RelA, NFκB1 and NFκB2 as well as IKKβ were associated with poor patient survival.
PI3K/AKT/mTOR/NFKB Axis
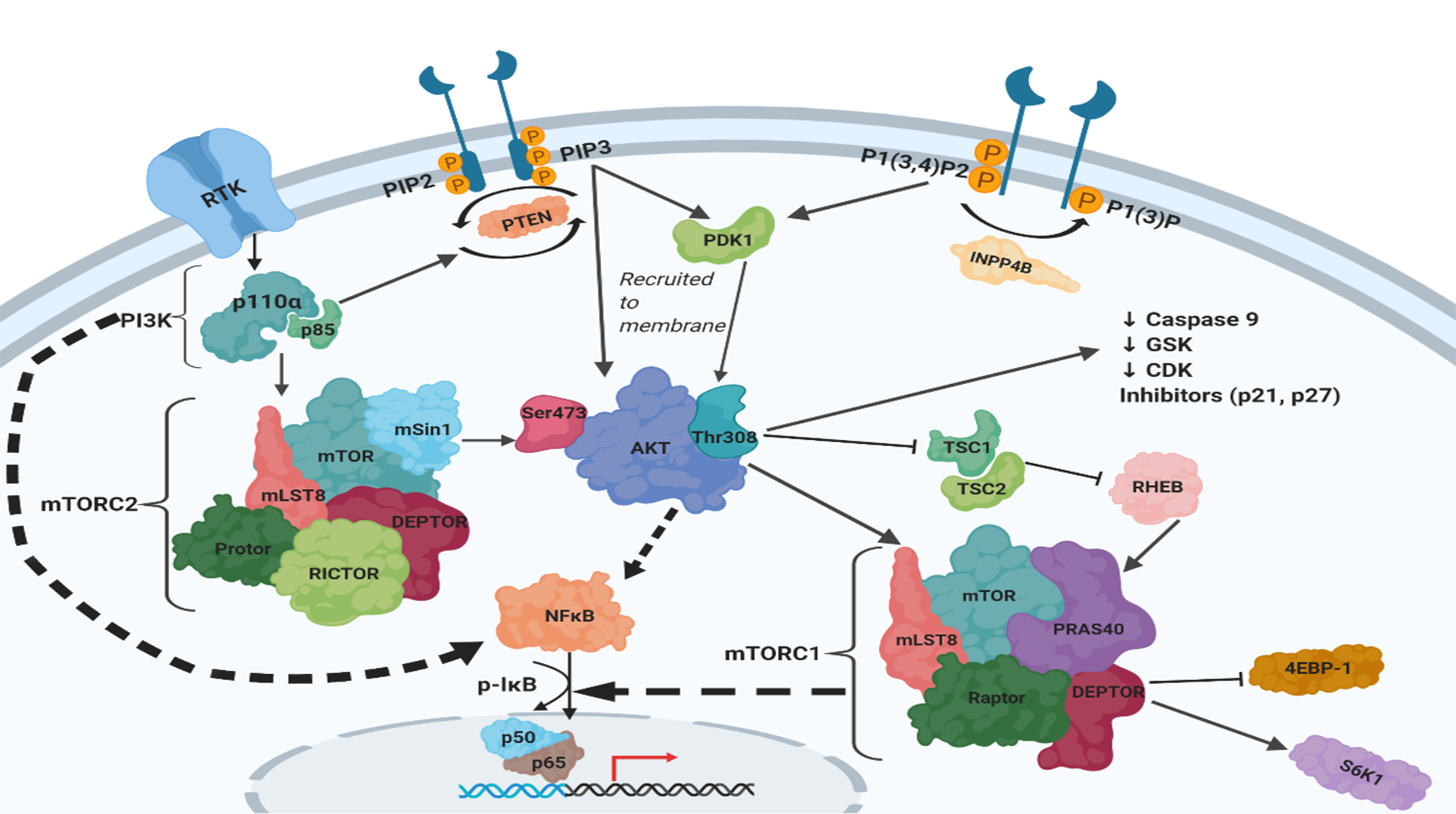
The PI3K and NFκB pathways are involved in a complex crosstalk (Figure 1) which results in decreased survival rates in OvCa patients [48]. The PI3K catalytic subunit p110α and its regulatory subunit p85 have been shown to directly activate NFκB [49-51]. Overexpression of the p110α subunit induces p65/RelA activation and nuclear translocation. PI3K activation also phosphorylates AKT with subsequent activation of the p65/RelA subunit of NFκB via phosphorylation through the IKK complex. Phospho-AKT mediates the phosphorylation of IKKα allowing for it to phosphorylate IkB, and hence allowing NFκB to translocate into the nucleus [52]. Moreover, AKT can activate NFκB independently of IKK by directly phosphorylating the p65/RelA subunit [53]. Importantly, analysis of TCGA data revealed a positive correlation between the transcripts of PIK3CA, AKT1/2/3, as well as NFkB subunits [28].
Recent Advances in Targeting PI3K/ AKT/mTOR/NFKB Axis
Several therapeutics are being developed in pre-clinical models to target PI3K/AKT/mTOR/NFκB axis in ovarian cancer. A seminal study by Yoon et al., recently reported that methyl lucidone (ML) from the dried fruit of Lindera erythrocarpa makino (Lauraceae) exerted cytotoxic effects in the OvCa cell lines, SKOV3 and OVCAR3. Specifically, ML inhibited cell proliferation with significant cellular morphological changes, and apoptosis in SKOV3. Mechanistically, ML induced apoptosis through cleavage of caspase-3/9 and Poly (ADP-Ribose) Polymerase (PARP), allowing for the release of cytochrome C from the mitochondria, decreased expression of Bcl-2 and BclxL, and prompted cell cycle arrest in the G2/M phase. ML also led to the repression of cyclin-A/B expression and stimulated cyclin-dependent kinase inhibitors p21 and p27. Importantly, ML exerted its inhibitory downstream effects by blocking the PI3K/AKT/mTOR/NFκB axis, manifested by significant downregulation of the levels of PI3K and phosphorylated AKT concomitant with nuclear translocation of NFκB and the total level of p-IkBα [54]. Another study reported that inhibition of YAP significantly suppressed the malignant behavior of OvCa cells, via regulation of the PI3K/AKT/mTOR pathway. Interestingly, a YAP inhibitor, peptide 17, inhibited OvCa progression by inhibiting the PI3K/AKT/mTOR pathway in vitro and in vivo [55]. In addition, Diaz- Cueto et al., [56] reported that pharmacologic inhibition of PI3K/AKT/mTOR, and ERK1/2 significantly reduced progranulin (PGRN) expression with subsequent inhibition of cell proliferation and survival in platinumresistant TOV-21G cells [56]. Interestingly, a recent study reported that PI3K/AKT/mTOR/NFκB axis is activated by ghrelin, an endogenous ligand for growth hormone secretagogue receptor (GHSR), promoting ovarian cancer cell survival and cisplatin resistance [57]. Targeting the ghrelin/ghrelin receptor pathway by ghrelin receptor antagonist, [D-Lys3]-GHRP-6 or the PI3K inhibitor, LY294002 significantly inhibited OvCa cell survival and sensitized them to cisplatin [57].
Clinically, a phase 2 clinical trial (NCT04055649), is ongoing using an orally active small molecule dopamine receptor D2 antagonist, ONC201, in combination with paclitaxel for the treatment of patients with platinum-resistant refractory or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer. ONC201 was originally identified as a small molecule that induces transcription of TNF-related apoptosisinducing ligand (TRAIL) and subsequently kills cancer cells by activating TRAIL death receptors [58]. Further investigation of the mechanism of action of ONC201 revealed that it acts through dual inhibition of AKT and ERK, [59], inhibition of NFκB and STAT3 [60] as well as inhibition of PI3K/AKT/mTOR [61] in a multitude of solid and hematologic malignancies, including ovarian cancer.
Acknowledgments
This work was funded by federal funds from the National Institutes of Health R01 CA193437 to N.S and T32 GM 127261 to A.G.
References
2. Mabuchi S, Kuroda H, Takahashi R, Sasano T. The PI3K/AKT/mTOR pathway as a therapeutic target in ovarian cancer. Gynecologic Oncology. 2015 Apr 1;137(1):173-9.
3. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nature Reviews Drug Discovery. 2014 Feb;13(2):140-56.
4. Marshall JD, Whitecross DE, Mellor P, Anderson DH. Impact of p85α alterations in cancer. Biomolecules. 2019 Jan;9(1):29.
5. Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nature Reviews Cancer. 2015 Jan;15(1):7-24.
6. Shayesteh L, Lu Y, Kuo WL, Baldocchi R, Godfrey T, Collins C, Pinkel D, Powell B, Mills GB, Gray JW. PIK3CA is implicated as an oncogene in ovarian cancer. Nature Genetics. 1999 Jan;21(1):99-102.
7. Kurman RJ, Shih IM. The dualistic model of ovarian carcinogenesis: revisited, revised, and expanded. The American Journal of Pathology. 2016 Apr 1;186(4):733- 47.
8. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011 Jun;474(7353):609.
9. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009; 458 (7239): 719–24.
10. Cai J, Xu L, Tang H, Yang Q, Yi X, Fang Y, Zhu Y, Wang Z. The role of the PTEN/PI3K/Akt pathway on prognosis in epithelial ovarian cancer: a meta-analysis.The Oncologist. 2014 May;19(5):528.
11. Carden CP, Stewart A, Thavasu P, Kipps E, Pope L, Crespo M, Miranda S, Attard G, Garrett MD, Clarke PA, Workman P. The association of PI3 kinase signaling and chemoresistance in advanced ovarian cancer. Molecular Cancer Therapeutics. 2012 Jul 1;11(7):1609-17.
12. Brasseur K, Gévry N, Asselin E. Chemoresistance and targeted therapies in ovarian and endometrial cancers. Oncotarget. 2017 Jan 17;8(3):4008.
13. Liu Z, Zhu G, Getzenberg RH, Veltri RW. The upregulation of PI3K/Akt and MAP kinase pathways is associated with resistance of microtubule-targeting drugs in prostate cancer. Journal of Cellular Biochemistry. 2015 Jul;116(7):1341-9.
14. Huang CZ, Wang YF, Zhang Y, Peng YM, Liu YX, Ma F, Jiang JH, Wang QD. Cepharanthine hydrochloride reverses P-glycoprotein-mediated multidrug resistance in human ovarian carcinoma A2780/Taxol cells by inhibiting the PI3K/Akt signaling pathway. Oncology Reports. 2017 Oct 1;38(4):2558-64.
15. Bumbaca B, Li W. Taxane resistance in castrationresistant prostate cancer: mechanisms and therapeutic strategies. Acta Pharmaceutica Sinica B. 2018 Jul 1;8(4):518-29.
16. Gasparri ML, Bardhi E, Ruscito I, Papadia A, Farooqi AA, Marchetti C, Bogani G, Ceccacci I, Mueller MD, Panici PB. PI3K/AKT/mTOR pathway in ovarian cancer treatment: are we on the right track?. Geburtshilfe und Frauenheilkunde. 2017 Oct;77(10):1095-103.
17. Martini M, De Santis MC, Braccini L, Gulluni F, Hirsch E. PI3K/AKT signaling pathway and cancer: an updated review. Annals of Medicine. 2014 Sep 1;46(6):372-83.
18. Spangle JM, Roberts TM, Zhao JJ. The emerging role of PI3K/AKT-mediated epigenetic regulation in cancer. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer. 2017 Aug 1;1868(1):123-31.
19. Li D, Pan Y, Huang Y, Zhang P, Fang X. Pak5 induces emt and promotes cell migration and invasion by activating the pi3k/akt pathway in ovarian cancer. Analytical Cellular Pathology. 2018;2018.
20. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007 Jun 29;129(7):1261- 74.
21. Mundi PS, Sachdev J, McCourt C, Kalinsky K. AKT in cancer: new molecular insights and advances in drug development. British Journal of Clinical Pharmacology. 2016 Oct;82(4):943-56.
22. Linnerth-Petrik NM, Santry LA, Moorehead R, Jücker M, Wootton SK, Petrik J. Akt isoform specific effects in ovarian cancer progression. Oncotarget. 2016 Nov 15;7(46):74820.
23. Cheaib B, Auguste A, Leary A. The PI3K/Akt/mTOR pathway in ovarian cancer: therapeutic opportunities and challenges. Chinese journal of cancer. 2015 Jan 1;34(1):4- 16.
24. Huang J, Manning BD. The TSC1–TSC2 complex: a molecular switchboard controlling cell growth. Biochemical Journal. 2008 Jun 1;412(2):179-90.
25. Qin X, Jiang B, Zhang Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle. 2016 Mar 18;15(6):781-6.
26. Hanrahan AJ, Schultz N, Westfal ML, Sakr RA, Giri DD, Scarperi S, Janikariman M, Olvera N, Stevens EV, She QB, Aghajanian C. Genomic complexity and AKT dependence in serous ovarian cancer. Cancer discovery. 2012 Jan 1;2(1):56-67.
27. Győrffy B, Lánczky A, Szállási Z. Implementing an online tool for genome-wide validation of survivalassociated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocrine-related Cancer. 2012 Apr 1;19(2):197-208.
28. Ghoneum A, Said N. PI3K-AKT-mTOR and NFκB Pathways in Ovarian Cancer: Implications for Targeted Therapeutics. Cancers. 2019 Jul;11(7):949.
29. Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. mTOR kinase structure, mechanism and regulation. Nature. 2013 May;497(7448):217-23.
30. Braunstein S, Karpisheva K, Pola C, Goldberg J, Hochman T, Yee H, Cangiarella J, Arju R, Formenti SC, Schneider RJ. A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer. Molecular Cell. 2007 Nov 9;28(3):501-12.
31. Beauchamp EM, Platanias LC. The evolution of the TOR pathway and its role in cancer. Oncogene. 2013 Aug;32(34):3923-32.
32. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006 Feb 10;124(3):471- 84.
33. Liu J, Wu DC, Qu LH, Liao HQ, Li MX. The role of mTOR in ovarian neoplasms, polycystic ovary syndrome, and ovarian aging. Clinical Anatomy. 2018 Sep;31(6):891- 8.
34. Altomare DA, Wang HQ, Skele KL, De Rienzo A, Klein-Szanto AJ, Godwin AK, Testa JR. AKT and mTOR phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene. 2004 Jul;23(34):5853-7.
35. Conciatori F, Ciuffreda L, Bazzichetto C, Falcone I, Pilotto S, Bria E, Cognetti F, Milella M. mTOR cross-talk in cancer and potential for combination therapy. Cancers. 2018 Jan;10(1):23.
36. Bai H, Li H, Li W, Gui T, Yang J, Cao D, Shen K. The PI3K/AKT/mTOR pathway is a potential predictor of distinct invasive and migratory capacities in human ovarian cancer cell lines. Oncotarget. 2015 Sep 22;6(28):25520.
37. Castellvi J, Garcia A, Rojo F, Ruiz-Marcellan C, Gil A, Baselga J, Ramon y Cajal S. Phosphorylated 4E binding protein 1: a hallmark of cell signaling that correlates with survival in ovarian cancer. Cancer. 2006 Oct 15;107(8):1801-11.
38. Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene, 2006. 25(51): 6680- 4.
39. Zheng C, Yin Q, Wu H. Structural studies of NF-κB signaling. Cell Research. 2011 Jan;21(1):183-95.
40. Hayden MS, Ghosh S. Shared principles in NF-κB signaling. Cell. 2008 Feb 8;132(3):344-62.
41. Karin M, Delhase M. The IκB kinase (IKK) and NF-κB: key elements of proinflammatory signalling. InSeminars in immunology 2000 Feb 1 (Vol. 12, No. 1, pp. 85-98). Academic Press.
42. Hunter JE, Leslie J, Perkins ND. c-Rel and its many roles in cancer: an old story with new twists. British Journal of Cancer. 2016 Jan;114(1):1-6.
43. Sun SC. The noncanonical NF-kappaB pathway. Immunology Reviews. 2012; 246(1): 125-40.
44. Jinawath N, Vasoontara C, Jinawath A, Fang X, Zhao K, Yap KL, Guo T, Lee CS, Wang W, Balgley BM, Davidson B. Oncoproteomic analysis reveals coupregulation of RELA and STAT5 in carboplatin resistant ovarian carcinoma. PloS one. 2010;5(6): e11198.
45. Ling L, Cao Z, Goeddel DV. NF-kappaB-inducing kinase activates IKK-alpha by phosphorylation of Ser- 176. Proceedings of the National Academy of Sciences of the United States of America, 1998. 95(7): 3792-7.
46. Kleinberg L, Dong HP, Holth A, Risberg B, Trope CG, Nesland JM, Flørenes VA, Davidson B. Cleaved caspase-3 and nuclear factor-κB p65 are prognostic factors in metastatic serous ovarian carcinoma. Human pathology. 2009 Jun 1;40(6):795-806.
47. Hernandez L, Hsu SC, Davidson B, Birrer MJ, Kohn EC, Annunziata CM. Activation of NF-kappaB signaling by inhibitor of NF-kappaB kinase beta increases aggressiveness of ovarian cancer. Cancer Research, 2010. 70(10): 4005-14.
48. Annunziata CM, Stavnes HT, Kleinberg L, Berner A, Hernandez LF, Birrer MJ, Steinberg SM, Davidson B, Kohn EC. Nuclear factor κB transcription factors are coexpressed and convey a poor outcome in ovarian cancer. Cancer. 2010 Jul 1;116(13):3276-84.
49. Sizemore N, Leung S, Stark GR. Activation of phosphatidylinositol 3-kinase in response to interleukin-1 leads to phosphorylation and activation of the NF-κB p65/RelA subunit. Molecular and Cellular Biology. 1999 Jul 1;19(7):4798-805.
50. Reddy SA, Huang JH, Liao WS. Phosphatidylinositol 3-Kinase In Interleukin 1 Signaling Physical Interaction with the Interleukin 1 Receptor and Requirement in NFκB and AP-1 Activation. Journal of Biological Chemistry. 1997 Nov 14;272(46):29167-73.
51. Koul D, Yao Y, Abbruzzese JL, Yung WA, Reddy SA. Tumor suppressor MMAC/PTEN inhibits cytokineinduced NFκB activation without interfering with the IκB degradation pathway. Journal of Biological Chemistry. 2001 Apr 6;276(14):11402-8.
52. Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-κB activation by tumour necrosis factor requires the Akt serine–threonine kinase. Nature. 1999 Sep;401(6748):82-5.
53. Madrid LV, Mayo MW, Reuther JY, Baldwin AS. Akt stimulates the transactivation potential of the RelA/ p65 subunit of NF-κB through utilization of the IκB kinase and activation of the mitogen-activated protein kinase p38. Journal of Biological Chemistry. 2001 Jun 1;276(22):18934-40.
54. Yoon JH, Shin JW, Pham TH, Choi YJ, Ryu HW, Oh SR, Oh JW, Yoon DY. Methyl lucidone induces apoptosis and G2/M phase arrest via the PI3K/Akt/NF-κB pathway in ovarian cancer cells. Pharmaceutical Biology. 2020 Jan 1;58(1):51-9.
55. Wei X, Jia Y, Lou H, Ma J, Huang Q, Meng Y, Sun C, Yang Z, Li X, Xu S, Yang X. Targeting YAP suppresses ovarian cancer progression through regulation of the PI3K/Akt/mTOR pathway. Oncology reports. 2019 Dec 1;42(6):2768-76.
56. Perez-Juarez CE, Arechavaleta-Velasco F, Zeferino- Toquero M, Alvarez-Arellano L, Estrada-Moscoso I, Diaz-Cueto L. Inhibition of PI3K/AKT/mTOR and MAPK signaling pathways decreases progranulin expression in ovarian clear cell carcinoma (OCCC) cell line: a potential biomarker for therapy response to signaling pathway inhibitors. Medical Oncology. 2020 Jan 1;37(1):4.
57. El-Kott AF, Shati AA, Al-Kahtani MA, Alqahtani S. Acylated Ghrelin Renders Chemosensitive Ovarian Cancer Cells Resistant to Cisplatin Chemotherapy via Activation of the PI3K/Akt/mTOR Survival Pathway. Analytical Cellular Pathology. 2019;2019: 9627810.
58. Allen JE, Kline CL, Prabhu VV, Wagner J, Ishizawa J, Madhukar N, Lev A, Baumeister M, Zhou L, Lulla A, Stogniew M. Discovery and clinical introduction of firstin- class imipridone ONC201. Oncotarget. 2016 Nov 8;7(45):74380.
59. Greer YE, Porat-Shliom N, Nagashima K, Stuelten C, Crooks D, Koparde VN, Gilbert SF, Islam C, Ubaldini A, Ji Y, Gattinoni L. ONC201 kills breast cancer cells in vitro by targeting mitochondria. Oncotarget. 2018 Apr 6;9(26):18454.
60. Ni X, Zhang X, Hu CH, Langridge T, Tarapore RS, Allen JE, Oster W, Duvic M. ONC201 selectively induces apoptosis in cutaneous T-cell lymphoma cells via activating pro-apoptotic integrated stress response and inactivating JAK/STAT and NF-κB pathways. Oncotarget. 2017 Sep 5;8(37):61761.
61. Feng Y, Zhou J, Li Z, Jiang Y, Zhou Y. Small molecular TRAIL inducer ONC201 induces death in lung cancer cells: a preclinical study. PloS one. 2016;11(9).