Plasmodium has a Non-canonical cAMP-regulated Pathway without G-proteins and GPCRs
Malaria is one of the most important disabling human, tropical disease caused by different Plasmodium species, which are protozoan parasites belonging to the Apicomplexa. The Apicomplexan parasites have a plastid like structure the “apicoplast” and comprise the genera Plasmodium, Toxoplasma and Cryptosporidium causing malaria, toxoplasmosis, and cryptosporidiosis. Despite enormous efforts and progress in drug discovery there is still a lack of drugs in the treatment of these neglected diseases mainly due to emerging resistance against commercialized drugs. In this view, an efficient hub on the identification of novel pathways harboring new drug targets is necessary. The fact that cAMP- regulated pathways in Apicomplexan parasites are unique makes them an attractive resource for drug discovery. The most important differences compared to the human host are summarized here in this commentary with a particular focus on Plasmodium.
In contrast to humans, the cyclic nucleotides cAMP and cGMP play an essential role in proliferation and differentiation which enables them to adapt to various environmental stimuli in the host cell. In canonical, cAMP/cGMP regulated pathways signal sensation is mediated through GPCRs (G-protein coupled receptors) [1]. Once a ligand has bound to a receptor it induces a conformational change of the receptor protein [2] which in turn leads to the binding of a heterotrimeric G-protein. Heterotrimeric G-proteins are a heterogenous group of proteins which consist of three different subunits, the α, β and χ subunits which interact with the receptor. Receptor activation leads to the exchange of GDP to GTP from the G-α subunit and in turn to the dissociation from the G-ßχ dimer. Both subunits are capable of independent signaling to downstream effectors like adenylyl cyclases or guanylcyclases which are responsible for cyclization of ATP/GTP to cAMP/cGMP. Once a threshold of both cyclic nucleotides has been reached, they are hydrolized by phosphodiesterases. Dependent on the cyclic nucleotide, either protein kinase A or cGMP-dependent protein kinase G is activated.
The most important, pathogenic parasite of the Apicomplexa is Plasmodium which causes malaria. After infection of the human host, the malaria parasite has to develop in different environments, i.e., the preerythrocytic stage in the human liver and the erythrocytic blood stages. Thereafter, a sexual state in the mosquito leads to the development of ookinetes in the midgut of the mosquito to form an oocyst which produces sporozoites in the salivary glands [3]. These developmental changes require a quick adaptation of the parasite that can only be achieved by signaling mechanisms. In contrast to research of cyclic nucleotide regulated pathways in humans research in Apicomplexan parasites has been delayed due to lack of available genome data. However, the recent sequencing results of their genomes revealed that their cyclic nucleotide pathways contain different components.
Surprisingly, canonical, heterotrimeric G-proteins and GPCRs are absent in the Apicomplexan parasites. Instead, in Plasmodium, a small set of Rab and Ras like GTPases is responsible for infection of the host cell [4]. Both belong to the Ras superfamily which has a variety of functions like gene expression control, cell proliferation, vesicle coating, nucleo-cytoplasmatic transport and regulation of cell cycle progression. Cycling occurs between an inactive GDP-bound form and an active GTPbound form which is controlled by activators like guanine nucleotide exchange factors (GEFs) and inhibitors i.e GTPase activating proteins (GAPs). Moreover, there is a strong interaction between host specific GTPases and GTPases from the parasite. Some of the host Rho GTPases can control innate and adaptive immune responses [5] while the parasite also can inactivate interferonregulated GTPases. Currently, 11 isoforms of Rabs have been identified in Plasmodium [6]. PfRab1a and PfRab1b regulate vesicular transport from the endoplasmic reticulum (ER) to the late- phase schizonts [7]. A unique, myristolated Rab5 GTPase has been characterized that is involved in the import of nutrients from hemoglobin of the infected host cell [8]. A recent screening for G-protein homologues in Plasmodium showed the presence of a Ras-like GTPase (acronym PfG) [9] with unknown function. PfG encodes a multi-enzyme-complex (109 kDa) and is localized to the cytosol. Collectively, targeting of parasite specific Rab GTPases might be a novel strategy in further drug development. Mammalian GPCRs belong to the group of the most commercialized drug targets with a potential in treatment of GPCRrelated disorders [10]. In contrast, four database entries of putative GPCRs currently exist in the Plasmodium database (PlasmoDB). Two of these putative GPCR sequences encode for serpentine receptors. One of them, the SR25 receptor protein, has been characterized as a monovalent cation sensor that modulates Ca2+ signaling in the cell. A decrease of high K+ concentration to low K+ concentration causes an increase in Ca2+ ions which can be reversed by blocking phospolipase C or depleting the parasite’s Ca2+ pool [11]. However, the druggability of the SR25 receptor protein is still in question since deletion experiments only showed insensitivity to hyperosmotic stress but did not eradicate the parasite [11]. Four cyclic nucleotide phosphodiesterases (PDEα-δ) have been identified in Plasmodium. Their sequence is highly conserved in comparison to the mammalian enzyme. Two of them i.e. PDEα and PDEß are expressed in the early erythrocytic stages, while the two others PDEγ and PDEδ are involved in sporozoite and gametocyte stages [12], respectively. PDEα is not essential in the blood stages but sensitive to the drug zaprinast. However, the IC50 value that was obtained was only in the μMol range which was later on improved using taldalafil and its derivatives [13] to increase the selectivity against the enzyme from the parasite.
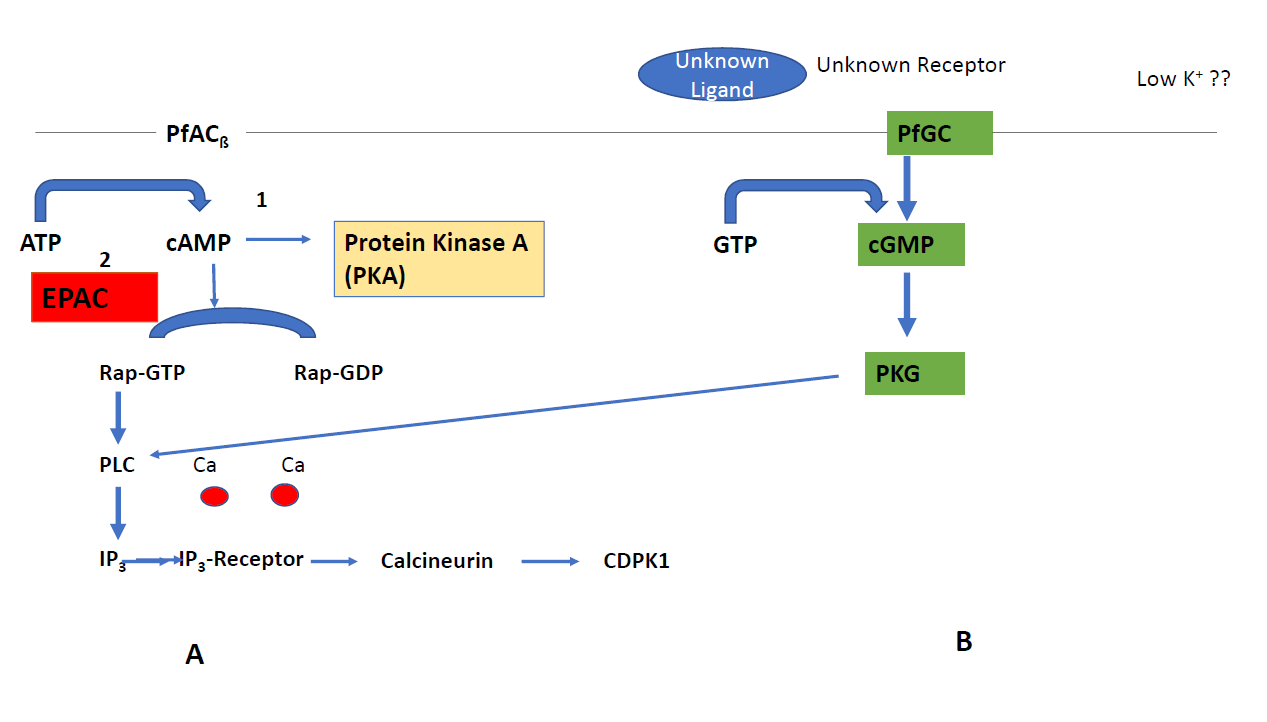
The most important peculiarity that appears in Plasmodium is the involvement of the guanine exchange factor (GEF) in the nucleotide exchange pathway (EPAC) and the rhoptry associated protein1 (Rap1) (Figure 1A) during egress and invasion of the parasite. One of the two adenylyl cyclases in Plasmodium, PfACß which is an orthologue of bicarbonate sensitive adenylyl cyclases, fosters the formation of cAMP. CAMP in turn activates the formation of Rap-GDP to Rap-GTP [14] triggering phospholipase C (PLC) to produce inositol triphosphate (IP3) that binds to the IP3 (IP3-R) receptor on the ER. This leads to a release in Ca-ions which bind to calcineurin and activation of Calcium dependent protein kinase 1 (CDPK1) that facilitates parasite invasion. Rap-1 has not been characterized in detail in Plasmodium although it might be an interesting drug candidate which controls a variety of effector proteins. Alternatively, cAMP can be used for signaling through protein kinase A (PKA)[15] (Figure 1A). Protein Kinase A from Plasmodium consists of two subunits. One subunit is catalytic (PKAc) while the other subunit has regulatory functions (PKAr). Once cAMP binds to (PKAr), PKAc is activated. In contrast to the mammalian host, PKAc lacks a dimerization domain responsible for the interaction with PKA activating proteins. PKA is essential in the asexual blood stages and acts at multiple stages in the parasite’s life cycle. The role of PKA in invasion into the human erythrocyte has also been shown [16]. These findings led to a search for inhibitors against PKA from the parasite. However, two inhibitors, i.e. H89 (5-isoquinolinesulfonamide) and the PKI inhibitor peptide (L-threonyl-L-threonyl-L-tyrosyl- L-alanyl-L-aspartyl-L-serylglycyl-phenylalanyl-L-isoleucyl- L-alanyl-L--arginyl-L-threonylglycyl-L-arginyl-Laspartic acid) exhibited a lower binding to the enzyme of the parasite in comparison to the human orthologue. Recently, a novel group of lead compounds, i. e., 3-methylisoquinoline-4 carbonitriles [17] showed more promising activity in Plasmodium in vitro cultures which is now further investigated with the purified enzyme.
A more attractive target is protein kinase G (PKG) since it is essential in all key stages of the parasite’s life cycle. PKG has several unique structural features: i) three cAMP/ cGMP binding motifs, ii) a degenerated cGMP binding site and iii) a lack of a leucine zipper motif responsible for dimerization. Moreover, PKG is insensitive to cGMP analogues. A capping mechanism by a pseudo-substrate covers the active site in the absence of cGMP. Mutations in the capping region are lethal in the asexual stages [18]. An inhibitor with an imidazopyridine lead structure [19] was identified which blocked the unusual gatekeeper position Thre618 in Plasmodium. Administration of this inhibitor resulted in reduced rounding up of gametocytes, ookinete gliding and prevention of schizont rupture. A second, important feature of PKG is the linkage to the nucleotide exchange pathway (EPAC) since it acts through CDPK1 (Figure 1B).
Toxoplasma gondii is Dependent on GPCR-mediated Signaling from the Infected Human Host Cell and a Set of its Own GTPases
Toxoplasmosis is caused by the ubiquitous protozoan parasite Toxoplasma gondii [20]. It infects 30% of the human population. During its life cycle it appears in three different developmental forms, i.e. the oocyste, the tachyzoite, and the bradyzoite. Transmission occurs through faeces from cats where the oocysts reside. The tachyzoite is the rapidly replicating form which leads to tissue damage and infection of the fetus in pregnant women which is called congenital toxoplasmosis. Dependent on the immune reaction of the human host, tachyzoites can develop into bradyzoites in particular in muscles and the central nervous system (CNS). However, some tachyzoites can escape the immune response of the host and develop back into bradyzoites. Bradyzoites can be ingested in meat and when taken up by the human host, they convert to tachyzoites.
Canonical G-proteins are also absent in Toxoplasma like in Plasmodium, instead a few proteins are found in the Toxoplasma database (http:/toxodb.org/toxo/) containing repeats of motifs of the ß-subunits of canonical G-proteins. No GPCRs occur in Toxoplasma except a database entry for a Rhodopsin-like transmembrane domain. Toxoplasma has a strong interaction with the GPCR-signaling network from its host [20]. Host cell cytolysis is induced by the parasite and involves two steps, i.e. ion loss and membrane poration [21]. However, recent data showed that host cell cytolysis is controlled by Gαq, phospholipase C (PLC), and protein kinase C (PKC). The mammalian PKC-inhibitor Go6976 (5,6,7,13-Tetrahydro- 13-methyl-5-oxo-12H-indolo[2,3-a]pyrrolo[3,4-c] carbazole-12-propanenitrile) limited T. gondii parasitic burden in vivo in spleen [22] of the infected mice. T. gondii lacks an orthologue of Rap-1, instead it interacts with immunity related GTPases (IRG). Rab GDP dissociation inhibitor α (RabGDIα) suppresses IFN-χ inducible host GTPases. Deficiency of RabGDIα resulted in enhanced IFN-χ mediated T. gondii clearance in vivo and in vitro [23].
The non-intracellular parasite Cryptosporidium proliferates without canonical G-proteins and GPCRs
Human cryptosporidiosis, a self-limiting diarrhea in healthy people is caused either by Cryptosporidium hominis or Cryptosporidium parvum [24] which can also infect animals. In immunodeficient people like HIV-1 infected persons or small children the disease can develop into a severe diarrhea with impact on the biliary tree and the respiratory organs [24]. The life cycle starts with the excretion of sporulated oocysts either by the faeces or by the respiratory organs. Following either ingestion or inhalation sporozoites are released and parasitize in epithelial cells. Then asexual multiplication starts (schizogeny or merogony) before microgamonts and macrogamonts are forming the oocyste in the sexual stage.
Due to a lack of canonical G-proteins and GPCRs Cryptosporidium ensures its own proliferation by a set of small GTPases. Notably, there is a database entry from Cryptosporidium muris encoding a developmentally regulated protein (DRG) protein 2 with 61% homology to the human paralogue [25]. DRGs are a family of highly conserved GTPases involved in eukaryotic translation [25]. A second, small, Ras-GTPase is expressed in cholangiocytes of the bile duct, mediating cytokine production and proliferation of the parasite after infection. Targeting this small Ras GTPase might be an alternative to eradicate Cryptosporidium infections. In sum, information on a cAMP-regulated pathway of this parasite is scarce.
Conclusions
In times of emerging resistance against conventional chemotherapy, the identification of pathways with novel targets is essential to control and eradicate important global, parasitic diseases. In this context, non-canonical cAMP-regulated pathways in Apicomplexan parasites causing malaria, toxoplasmosis and cryptosporidiosis provide a valuable tool. This commentary shows different strategies how to tackle the problem.
1. Since canonical G-proteins and GPCRs are absent in these parasites but present in the mammalian host, inhibition of the host proteins might reduce parasite load. All three genera of the Apicomplexan parasites are strongly dependent on canonical, cAMP-regulated pathways of the human host.
2. Inhibition of the Rap1 protein in the guanine nucleotide exchange pathway would be an attractive way to prevent invasion and egression of the parasite.
3. From the secondary effector proteins, i.e. the family of ACG kinases, kinase G is the most promising target in Plasmodium since it has regulatory key functions in all stages of the parasite’s life cycle. An inhibitor with imidazopyridine lead structure [19] already fulfills the WHO’s requirement that a single drug eradicates the parasite in each developmental stage.
4. Current drug discovery has not exploited small Ras GTPases in all three genera which interact with specific host enzymes to accelerate invasion and proliferation of the parasite.
References
2. Baltoumas FA, Theodoropoulou MC, Hamodrakas SJ. Interactions of the α-subunits of heterotrimeric G-proteins with GPCRs, effectors and RGS proteins: a critical review and analysis of interacting surfaces, conformational shifts, structural diversity and electrostatic potentials. Journal of Structural Biology. 2013 Jun 1;182(3):209-18.
3. Zheng W, Liu F, He Y, Liu Q, Humphreys GB, Tsuboi T, et al. Functional characterization of Plasmodium berghei PSOP25 during ookinete development and as a malaria transmission-blocking vaccine candidate. Parasites & Vectors. 2017 Dec;10(1):8.
4. Rojas AM, Fuentes G, Rausell A, Valencia A. The Ras protein superfamily: evolutionary tree and role of conserved amino acids. Journal of Cell Biology. 2012 Jan 23;196(2):189-201.
5. Thumkeo D, Watanabe S, Narumiya S. Physiological roles of Rho and Rho effectors in mammals. European Journal of Cell Biology. 2013 Oct 1;92(10-11):303-15.
6. Zhang P, Xiao Z, Wang S, Zhang M, Wei Y, Hang Q, et al. ZRANB1 Is an EZH2 Deubiquitinase and a Potential Therapeutic Target in Breast Cancer. Cell reports. 2018 Apr 17; 23(3):823-37.
7. Morse D, Webster W, Kalanon M, Langsley G, McFadden GI. Plasmodium falciparum Rab1A localizes to rhoptries in schizonts. PloS One. 2016;11(6).
8. Ezougou CN, Ben-Rached F, Moss DK, Lin JW, Black S, Knuepfer E, et al. Plasmodium falciparum Rab5B is an N-terminally myristoylated Rab GTPase that is targeted to the parasite’s plasma and food vacuole membranes. PloS One. 2014;9(2).
9. Kaiser A, Langer B, Przyborski J, Kersting D, Krüger M. A putative non-canonical Ras-like GTPase from P. falciparum: chemical properties and characterization of the protein. PloS One. 2015;10(11).
10. Ayoub MA. Small molecules targeting heterotrimeric G proteins. European Journal of Pharmacology. 2018 May 5;826:169-78.
11. Moraes MS, Budu A, Singh MK, Borges-Pereira L, Levano-Garcia J, Currà C, et al. Plasmodium falciparum GPCR-like receptor SR25 mediates extracellular K+ sensing coupled to Ca 2+ signaling and stress survival. Scientific Reports. 2017 Aug 25;7(1):1-3.
12. Yuasa K, Mi-Ichi F, Kobayashi T, Yamanouchi M, Kotera J, Kita K, Omori K. PfPDE1, a novel cGMP-specific phosphodiesterase from the human malaria parasite Plasmodium falciparum. Biochemical Journal. 2005 Nov 15;392(1):221-9.
13. Beghyn TB, Charton J, Leroux F, Henninot A, Reboule I, Cos P, et al. Drug-to-genome-to-drug, step 2: reversing selectivity in a series of antiplasmodial compounds. Journal of Medicinal Chemistry. 2012 Feb 9;55(3):1274-86.
14. Soni R, Sharma D, Rai P, Sharma B, Bhatt TK. Signaling Strategies of Malaria Parasite for its Survival, Proliferation, and infection during erythrocytic Stage. Frontiers in Immunology. 2017 Mar 28;8:349.
15. Ramdani G, Naissant B, Thompson E, Breil F, Lorthiois A, Dupuy F, et al. cAMP-signalling regulates gametocyte-infected erythrocyte deformability required for malaria parasite transmission. PLoS Pathogens. 2015 May;11(5).
16. Leykauf K, Treeck M, Gilson PR, Nebl T, Braulke T, Cowman AF, Gilberger TW, Crabb BS. Protein kinase a dependent phosphorylation of apical membrane antigen 1 plays an important role in erythrocyte invasion by the malaria parasite. PLoS Pathogens. 2010 Jun;6(6):e1000941.
17. Buskes MJ, Harvey KL, Prinz B, Crabb BS, Gilson PR, Wilson DJ, Abbott BM. Exploration of 3-methylisoquinoline-4-carbonitriles as protein kinase A inhibitors of Plasmodium falciparum. Bioorganic & Medicinal Chemistry. 2016 Jun 1;24(11):2389-96.
18. Gurnett AM, Liberator PA, Dulski PM, Salowe SP, Donald RG, Anderson JW, Wiltsie J, Diaz CA, Harris G, Chang B, Darkin-Rattray SJ. Purification and molecular characterization of cGMP-dependent protein kinase from apicomplexan parasites a novel chemotherapeutic target. Journal of Biological Chemistry. 2002 May 3;277(18):15913-22.
19. Green JL, Moon RW, Whalley D, Bowyer PW, Wallace C, Rochani A, et al. Imidazopyridazine inhibitors of Plasmodium falciparum calcium-dependent protein kinase 1 also target cyclic GMP-dependent protein kinase and heat shock protein 90 to kill the parasite at different stages of intracellular development. Antimicrobial Agents and Chemotherapy. 2016 Mar 1;60(3):1464-75.
20. Millholland MG, Mishra S, Dupont CD, Love MS, Patel B, Shilling D, Kazanietz MG, Foskett JK, Hunter CA, Sinnis P, Greenbaum DC. A host GPCR signaling network required for the cytolysis of infected cells facilitates release of apicomplexan parasites. Cell Host & Microbe. 2013 Jan 16;13(1):15-28.
21. Moudy R, Manning TJ, Beckers CJ. The loss of cytoplasmic potassium upon host cell breakdown triggers egress of Toxoplasma gondii. Journal of Biological Chemistry. 2001 Nov 2;276(44):41492-501.
22. Kremer K, Kamin D, Rittweger E, Wilkes J, Flammer H, Mahler S, et al. An overexpression screen of Toxoplasma gondii Rab-GTPases reveals distinct transport routes to the micronemes. PLoS Pathogens. 2013 Mar;9(3).
23. Ohshima J, Sasai M, Liu J, Yamashita K, Ma JS, Lee Y, Bando H, Howard JC, Ebisu S, Hayashi M, Takeda K. RabGDIα is a negative regulator of interferon-γ– inducible GTPase-dependent cell-autonomous immunity to Toxoplasma gondii. Proceedings of the National Academy of Sciences. 2015 Aug 18;112(33):E4581-90.
24. Leitch GJ, He Q. Cryptosporidiosis-an overview. Journal of Biomedical Research. 2011 Jan 1;25(1):1-6.
25. Francis SM, Gas ME, Daugeron MC, Bravo J, Seraphin B. Rbg1–Tma46 dimer structure reveals new functional domains and their role in polysome recruitment. Nucleic Acids Research. 2012 Nov 1;40(21):11100-14.