Abstract
Cullin-RING E3 ubiquitin ligase 4 (CRL4) plays an essential role in cell cycle progression. Recent efforts using high throughput screening and follow up hit-to-lead studies have led to identification of small molecules 33-11 and KH-4-43 that inhibit E3 CRL4’s core ligase complex and exhibit anticancer potential. This review provides: 1) an updated perspective of E3 CRL4, including structural organization, major substrate targets and role in cancer; 2) a discussion of the challenges and strategies for finding the CRL inhibitor; and 3) a summary of the properties of the identified CRL4 inhibitors as well as a perspective on their potential utility to probe CRL4 biology and act as therapeutic agents.
Keywords
E3 CRL4, Cdt1, Cell cycle, Small molecule inhibitors, Tumor inhibition
Introduction
Cullin-RING (Really Interesting New Gene) E3 ubiquitin (Ub) ligases (CRLs) are RING-type E3s characterized by a signature Cullin-RING heterodimeric complex [1,2]. There are six canonical cullin (CUL) proteins, CUL1, CUL2, CUL3, CUL4A, CUL4B, and CUL5, that all adopt an elongated structure to function as a scaffold for assembly of E3 CRL. Typically, a CUL’s N-terminal domain assembles interchangeably with CUL-specific substrate receptors capable of binding a substrate. On the other hand, a CUL’s C-terminal domain (CTD) binds a RING finger protein, ROC1/RBX1 for CUL1 to 4 or ROC2 for CUL5, to form a core ligase complex. This modular feature allows for assembly of a diverse set of structurally similar, but functionally distinct CRLs. There are ~300 CRL members in humans, comprising ~50% of the E3s identified to date [1,2]. This review article focuses on E3 CRL4 and highlights recent developments aimed at targeting this E3 by small molecule inhibitors.
E3 CRL4: Structural Organization
The resolved crystal structure of CRL4ASV5-V [3,4] provides an example of the overall architecture of E3 CRL4. It contains the CUL4A scaffold, the 127 kDa DNA damage-binding protein 1 (DDB1) as an adaptor, SV5-V that is a substrate receptor that belongs to the DDB1– cullin-4-associated factor (DCAF; ~90 in humans) protein family, and ROC1/RBx1 RING finger protein (Figure 1). DDB1 utilizes three β-propellers, BPA, BPB, and BPC, and a C-terminal helical domain for interactions with the N-terminal domain (NTD) of CUL4A, as well as a DCAF protein such as SV5-V, respectively. Intriguingly, comparison of a number of DDB1 structures resolved to date has revealed a significant level of plasticity, suggesting an ability of DDB1 to adopt flexible orientations that might enable E3 CRL4 to optimally position a DCAF for substrate ubiquitination, and/or accommodate substrate polyubiquitination [5].
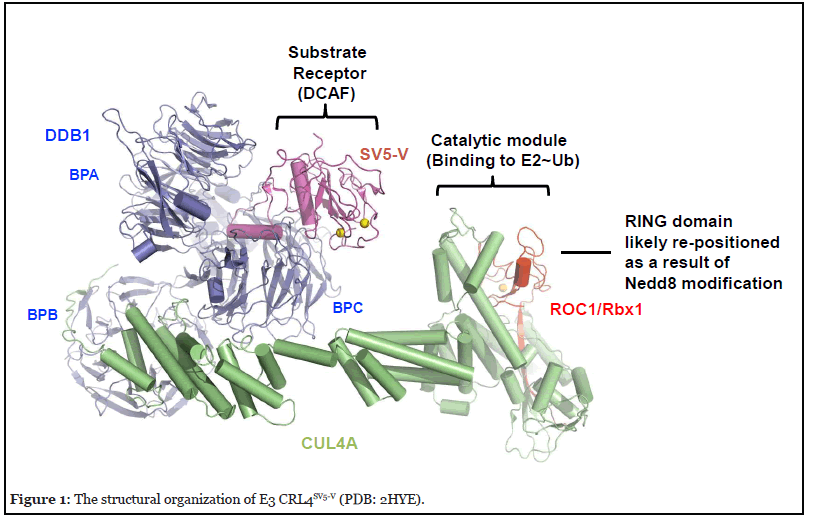
At its C-terminus CUL4A interacts with ROC1/Rbx1 via both “strong” and “weak” modes. The “strong” interactions involve ROC1/RBX1’s β-strand that inserts itself between multiple CUL4A’s β-strands, creating a stable intermolecular β-sheet structure. By contrast, the rest of CUL4 C-terminal domain (CTD) appears to engage with the RING domain of ROC1/RBX1 through a loose interface. Biochemical studies using the CUL1-based CRL complexes suggest that part of the cullin-ROC1 RING domain interface interactions acts to restrain the E3-mediated ubiquitination activity and that such autoinhibition is reversed by the action of Nedd8 [6]. Nedd8 is a ubiquitinlike protein that is covalently conjugated to a conserved residue within the cullin CTD [7]. Remarkably, conjugation of CUL5 by Nedd8 was shown to cause the ROC1 RING domain to pop out [8]. This re-positioning of ROC1 RING finger likely establishes an open active conformation optimal for interactions with an E3-bound substrate and an E2~ubiquitin thiol ester, resulting in the transfer of ubiquitin to the substrate.
The E3 CRL4DCAF2-Cdt1 Pathway and CRL4CRBN
Targeted degradation of Cdt1 (CDC10-dependent transcript 1) by E3 CRL4DCAF2 [9-11] is one of the best characterized evolutionarily conserved proteolytic mechanisms (Figure 2). Cdt1 is a DNA replication initiation factor that together with Cdc6 (cell division cycle 6), functions to load the MCM (mini-chromosome maintenance protein complex) DNA replicative helicase near to sites of replication origin. As such, Cdt1 is widely considered as a licensing factor because its action leads to replication origin unwinding and then replicative DNA synthesis [12,13]. Prompt removal of Cdt1 after origin firing by E3 CRL4DCAF2 and the proteasome is critical to prevent re-replication and thus ensure replication only once per cell cycle [14,15]. Of particular importance, DCAF2 (also called Cdt2) is responsible for targeting Cdt1 in a manner that requires DNA and PCNA (proliferating cell nuclear antigen). While the DCAF2/Cdt1/PCNA/DNA interactions appear complex and a detailed mechanism remains elusive, it is clear that a conserved PCNA motif known as PCNA-interacting protein (PIP) box plays a significant role [16-19].
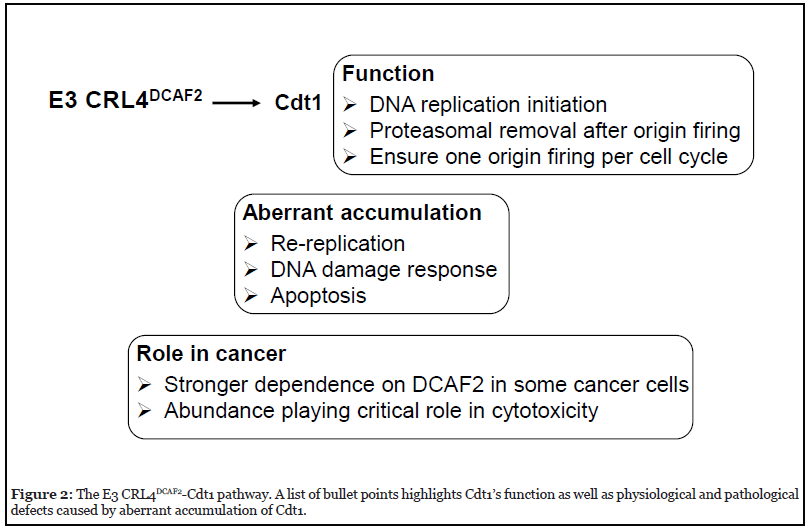
The CRL4DCAF2-directed Cdt1 turnover is of fundamental importance to cell cycle progression (Figure 2). The Kipreos group first observed massive DNA re-replication with elevated DNA levels up to 100C content driven by aberrant accumulation of Cdt1 in Caenorhabditis elegans lacking CUL4[14]. Similar defects have been observed in mammals. For example, mouse embryos derived from dcaf2-null oocytes were found to arrest at the one- to two-cell stage, owing to prolonged DNA replication and accumulation of massive DNA damage [20]. In addition, defects observed in the context of dcaf2 deletion were phenocopied by over-expression of Cdt1 in wild type cells, supporting the notion that Cdt1 abundance is a critical determinant for cell cycle progression. As revealed by multiple studies [21-24], apoptosis is a frequently observed cellular response to DNA damage triggered by Cdt1 accumulation. The Cdt1-CRL4DCAF2 pathway appears to participate in DNA repair/checkpoint control. Upon exposure to UV or ionizing radiation, Cdt1 is rapidly ubiquitinated and degraded by CRL4Cdt2 to halt DNA replication origin firing [11,17,25-26].
CRL4DCAF2 has several additional substrates. It targets the degradation of cyclin-dependent kinase inhibitor p21 [27-29], and histone H4 methyltransferase Set8 [30,31]. Their reaction mechanisms may resemble that of Cdt1 because they share the same requirement for PCNA. Redundant E3s for p21 degradation have been reported. For example, the Kipreos group showed that in asynchronously growing HeLa cells, knockdown of Skp2 and DCAF2 each resulted in accumulation of p21, while the combined knockdown produced the most pronounced cooperative effect [28].
CRL4CRBN has received significant attention in recent years because the DCAF protein cereblon (CRBN) was identified as a target bound to the Food and Drug Administration (FDA)-approved anticancer drug class known as Immunomodulatory imide drugs (IMiDs) that include thalidomide, lenalidomide, pomalidomide, etc [32]. Thanks to a large number of elegant genetic, biochemical and structure studies, it is now widely accepted that binding of an iMiD to CRBN creates new protein-protein interaction surface, thus enabling the iMiD-bound E3 to target a novel set of neo-substrate proteins that include IKZF1/3 (Ikaros family zinc finger protein 1/3), CK1α (casein kinase 1α), and GSPT1 (G1-to-S phase transition 1) [33-38]. Owing to the high affinity of iMiDs for CRBN, CRL4CRBN has become a widely used and highly effective platform for PROTACs (proteolysis targeting chimeras), in which a dual-functional small molecule degrader can be modularly designed to contain both an iMiD and a ligand with affinity for a selected specific cellular protein targeted for degradation [39]. Once introduced into cells, the degrader is able to harness its desired targeted cellular protein substrate to E3 CRL4CRBN for ubiquitination and degradation. Notably, the iMiDs-dependent ubiquitination of a neo-substrate (such as CK1α) by CRL4CRBN has been reconstituted in vitro [36,37,40], creating tools that enable in-depth biochemical mechanistic studies and drug testing efforts. Intriguingly, a similar drug-harnessing mechanism appears to be utilized by another DCAF protein, DCAF15, that is bound to sulfonamides which then gain the ability to target RBM39 for degradation [41].
Role of E3 CRL4 in Cancer
CUL4A and CUL4B are highly similar except that CUL4B contains a distinct N-terminal extension that includes a sequence encoding for a nuclear localization signal, thereby making it a predominantly nuclear protein [42]. CUL4A and CUL4B are broadly co-expressed. Multiple studies suggest no overt growth abnormalities in germline Cul4a or Cul4b knockout mice [43-45]. However, deletion of CUL4A and CUL4B in mouse embryonic fibroblasts, as well as cultured tumor cells, results in growth retardation [43], suggesting that the CRL4 E3 ligase activity plays an essential role in growth. Genetic ablation of the Ddb1, the adaptor for both CUL4A and CUL4B, is embryonic lethal [46-47]. The above findings collectively suggest that E3 CRL4 is essential for murine development, and that CRL4A and CRL4B are functionally redundant.
Previous studies have suggested the importance of both CUL4A and CUL4B in cancer [48]. Studies in mice using either overexpression or silencing approaches indicate that both CUL4A and CUL4B play a tumor-promoting role in many cancer types including lung [49,50], breast [51], colon [52,53], and hepatocellular carcinomas [54-55]. In addition, CUL4 overexpression has been linked to an array of human cancers including tumors of the breast [56], ovary [57], stomach [58-59], colon [51-53, 60], pancreas [61], lung [62] and bile duct [63-65].
Consistent with its essential role in cell cycle progression by targeting Cdt1 for degradation, deletion of the dcaf2 gene in mice causes early embryonic lethality [66]. Elevated expression of DCAF2 is detected in ovarian cancer [21], melanoma [67], Ewing sarcoma [68], and head and neck cancer [69]. Previous studies suggest a role of the CDT1/CRL4DCAF2 pathway in tumorigenesis. First, it was shown that RNA interference (RNAi)-mediated depletion of DCAF2 caused apoptosis in the 12 tumor cell lines examined, but not in 6 non-transformed cell lines, suggesting that cancer cells are dependent on the functional targeting of Cdt1 by CRL4DCAF2 at levels much higher than normal cells [70]. Moreover, Cdt1 knockdown was able to partially reduce apoptosis in cells in which all E3 CRL activities were neutralized by the Nedd8 inhibitor MLN4924/Pevonedistat, suggesting a significant role for Cdt1 abundance in mediating genotoxic effects of the anticancer drug [21] (Figure 2).
Targeting E3 CRL: Challenge and Strategy
A selective small-molecule modulator of CRLs’ function would facilitate mechanistic and phenotypic studies into CRL functions and provide a tool to identify their targets in biochemical, cell-based, and animal studies. To date, there is only one FDA-approved E3 drug class that targets the DCAF substrate receptor CRBN (thalidomide/ lenalidomide) [71]. Current drug/probe discovery efforts against the ubiquitin-proteasome system depend heavily on traditional methods that exploit the ability of smallmolecule agents to disable an enzyme’s catalytic pocket. Examples include USP7 inhibitors P50429/P22077 (modifying the de-ubiquitinating enzyme [DUB]’s catalytic cysteine residue) [72], Nedd8 inhibitor MLN4924 (forming a covalent adduct with Nedd8 to bind and inactivate Nedd8 E1 enzyme) [73,74], and proteasome drug Bortezomib (binding to, and inhibiting, the protease’s catalytic site) [75].
However, RING-type E3s are atypical enzymes that contribute to ubiquitination by mediating protein–protein interactions (PPI) with substrate, E2, and Ub [1]. Previous successful PPI drugs typically exploit a deep hydrophobic pocket in a target. For example, E3 MDM2 (mouse double minute 2) has a deep hydrophobic cavity formed by 14 amino acids, with which three amino acids from p53 directly interact [76]. Several small molecule inhibitors including nutlins [77; reviewed by ref. 78], have been developed and their mechanism of action is to occupy the MDM2 pocket and displace p53, thereby protecting p53 from being degraded by MDM2. However, high-resolution structural studies have shown that interactions involving E2’s RING domain, E2, and Ub are characterized by large, relatively flat interfaces [79]. Such perceived “undruggable” features impose a significant barrier to utilize structure-based ligand search using either virtual docking or fragment based physical screening for the discovery of CRL RING domain PPI drugs.
To address the need to find a chemical probe for E3 CRL’s core ligase complex, a high-throughput screening (HTS) platform was created using the fluorescence (Förster) resonance energy transfer (FRET) K48 di-Ub assay (80- 81) (Figure 3A). In this system, incubation of E1, E2 Cdc34, and an E3 CRL1 subcomplex (ROC1–CUL1 CTD) results in covalent conjugation of the donor Ub to the receptor Ub. Because the donor and receptor ubiquitins carry a pair of matching fluorophores at specific sites, the conjugation of these two molecules allows for optimal FRET energy transfer from the excited donor fluorophore to the receptor fluorophore, producing a distinct fluorescence signal.
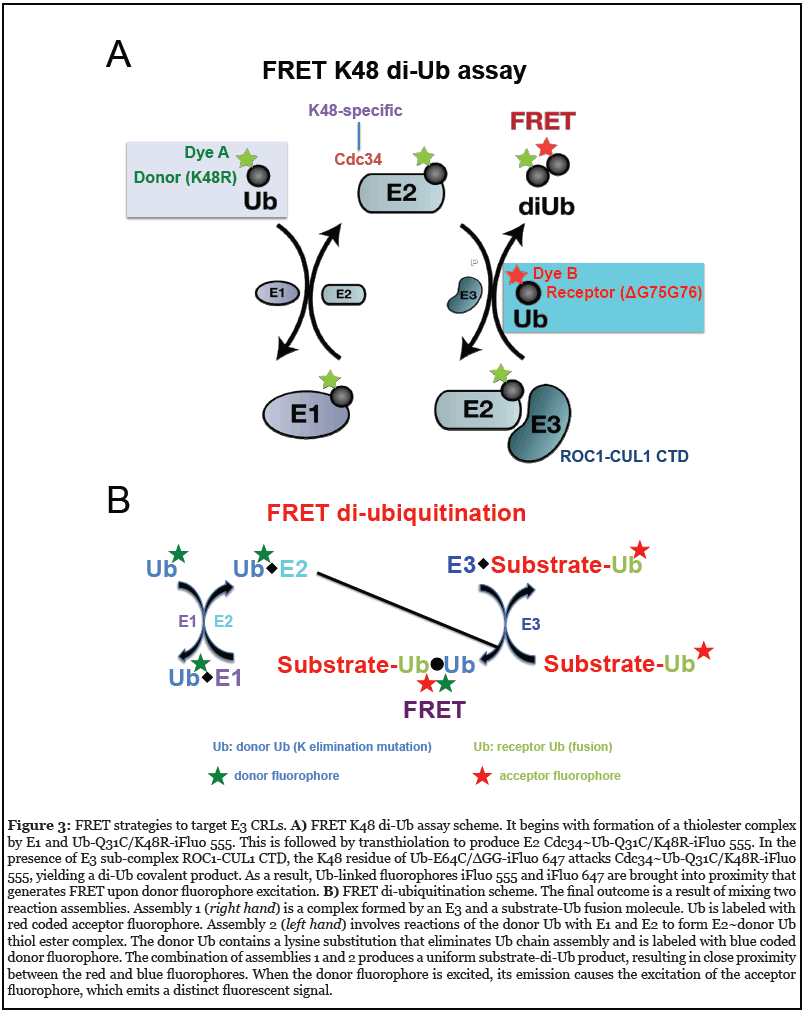
Fully functional Ub variants were employed to allow only one nucleophilic attack. As a consequence, a single Ub– Ub isopeptide bond is produced, thereby eliminating the complexity associated with polyubiquitin chain assembly which ensures a high degree of reproducibility for effective HTS. A pilot HTS identified a small molecule compound, suramin (an anti-trypanosomal drug), that can inhibit E3 CRL1 activity by disrupting its ability to recruit E2 Cdc34 [80]. These results have provided proof-of-principle evidence that an E2–E3 interface can be perturbed with small molecule modulators. Using this approach, largescale HTS with extensive follow-up hit-to-lead studies, led to identification of a class of small-molecule inhibitors against E3 CRL4 [82].
More recently, a FRET diubiquitination assay (Figure 3B) was developed to track substrate ubiquitination by fluorescence [83]. While FRET diubiquitination is derived from the FRET K48 di-Ub assay (Figure 3A), it requires E3-substrate interactions. Hence this new reporter system could be used in the future HTS campaigns to discover small-molecule modulators capable of targeting specific substrate–E3 interfaces.
Discovery of Inhibitors against E3 CRL4
In an effort to discover small molecule modulators that target E3 CRL, HTS and follow up hit-to-lead studies were carried out, resulting in identification of two compounds, 33-11 and KH-4-43, which inhibit E3 CRL4 and exhibit antitumor potential [82]. These compounds bind to CRL4’s core catalytic complex, inhibit E3 CRL4ACRBN-dependent ubiquitination of CK1α in vitro, and cause stabilization of CRL4’s substrate Cdt1 in cells (Figure 4). Treatment with 33-11 or KH-4-43 in a panel of 36 tumor cell lines revealed cytotoxicity, which was linked to aberrant accumulation of Cdt1 known to trigger apoptosis.
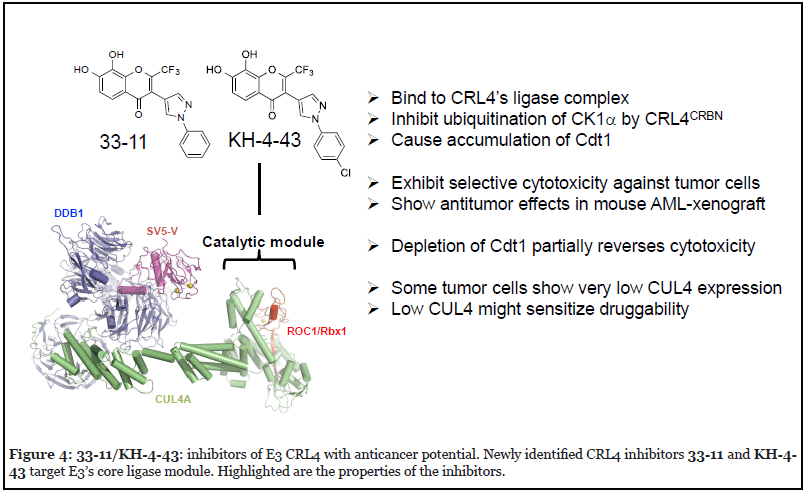
According to the Structural Genomics Consortium, chemical probes are required to minimally have in vitro potency of the target protein at <100 nM, possess >30X selectivity relative to other sequence-related proteins of the same target family, and have demonstrated on-target effects at <1 μM [84]. KH-4-43 appears close to these criteria because it has a binding Kd to E3 ROC1–CUL4A CTD or the highly related ROC1–CUL1 CTD at 83 nM or 9.4 μM, respectively [82], which represents a difference of two orders of magnitude. KH-4-43 exhibits cytotoxicity in a subset of tumor cell lines with EC50 approaching ~2 μM. The on-target effects were supported by the results of RNAi experiments, which demonstrated that 33-11‘s cytotoxicity was enhanced by CUL4 depletion, but was partially overcome by knockdown of CDT1.
Comparison of KH-4-43 and 33-11 revealed improved potency for KH-4-43 as an inhibitor of E3 CRL4 [82].KH-4-43 is more effective than 33-11 in binding to CRL4’s core ligase subcomplex, inhibiting the ubiquitination of CK1α by E3 CRL4CRBN in vitro, and stabilizing CRL4’s substrate CDT1 in cells. KH-4-43 exhibits higher levels of cytotoxicity and in vivo antitumor activity than 33-11.
Thus, it appears possible for a small molecule agent to selectively target a specific cullin CTD. Despite a common globular CTD adopted by cullins 1 to 5, however, different cullin CTDs have divergent folds [4,8,85,86] and different total areas of interface with ROC1/Rbx1 that result in significantly divergent orientation of the ROC1/Rbx1 RING domain among CRLs [86]. It is therefore conceivable that a small molecule (such as 33-11/KH-4-43) may bind to a specific site within a cullin CTD that impacts the cullin– E2 interaction and/or alters the ROC1/Rbx1 orientation, leading to selective inhibition of ubiquitination.
The 33-11/KH-4-43 class of inhibitors may prove useful as tools for probing CRL-mediated biological signaling pathways and mechanisms. A co-crystal structure of E3 CRL4 with 33-11/KH-4-43 (or improved analog) will reveal critical insights into the ligand-target interactions to guide structure-based design of compounds to improve inhibitory potency and selectivity. Moreover, by determining the binding mode of 33-11/KH-4-43 on CRL4, such studies may reveal how ligand-E3 interactions might inhibit ubiquitination, hence providing fresh insights for the use of chemical biology strategies to modulate CRL activity in a manner that broadly impacts the ubiquitin-proteasome field. Proteomic approaches using 33-11/KH-4-43 may identify cellular targets that discover new E3-mediated biological interactions and pathways. For example, the Ebert group has employed a SILAC-based global search of protein abundance changes in cells treated with lenalidomide, which helped identify substrate CK1α [36].
Based on preliminary structure activity relationship (SAR) studies, the 33-11/KH-4-43 scaffolds are highly amenable to medicinal chemistry optimization with several substituents available for modification and building blocks that have been easily incorporated into the scaffold [82]. Continued characterization of the key pharmacophores of validated leads will provide improved CRL4 potency and selectivity, as well as drug-like properties and pharmacokinetic parameters.
Inhibitors of E3 CRL4 Have Anticancer Potential
Inhibitors of E3 CRL4 33-11/KH-4-43 are toxic to a panel of tumor cells and such cytotoxicity can be partially reversed by depletion of Cdt1 [82]. These findings suggest that aberrant accumulation of CDT1, as a result of inactivation of E3 CRL4 by 33-11/KH-4-43, triggers apoptosis in some cancer cells. Intriguingly, a subset of tumor cells was found to express CUL4 proteins at levels as much as 70-fold lower than those in other tumor lines [82]. In a strong support of the key role played by CUL4 abundance in these drug effects, it was shown that reducing CUL4 levels by means of siRNA-mediated depletion sensitized U2OS cells to 33-11 treatment for apoptosis [82]. 33-11/KH-4-43 exhibited in vivo anti-tumor activity in an Acute Myelogenous Leukemia (AML) xenograft mouse model study [82]. Taking together, these findings suggest that 33-11/KH-4-43-based CRL4 inhibitors may provide new exploitable therapeutic opportunities to target against a subset of leukemias that are characterized by low CUL4 expression (Figure 4).
The major impact of low target expression in enhancing druggability has been previously recognized as clinically significant. A well-known example, leukemic del(5q) myelodysplastic syndrome cells, are haplo-insufficient for CK1α and these cells are sensitized to lenalidomide therapy that specifically targets CK1α for degradation [36]. If validated, the low-target expression driven drug sensitivity mechanism may be advantageous as compared with targeting amplified or mutated oncoproteins because targeting these oncogenic drivers frequently leads to selection events such as secondary mutations which result in drug resistance [87].
Concluding Remarks
Targeting the ubiquitin-proteasome system is a new frontier for drug discovery [71]. E3 CRLs play a critical role in protein homeostasis and cell maintenance. Their functions are critical for tumor cell survival making them potentially actionable targets for cancer therapy. Through the development of assays and tools that allow us to interrogate the function and requirements for specific CRLs in normal and tumor cells we can gain insight into tumor specific dependencies upon specific CRLs and develop agents that can specifically modulate their activities.
An overwhelming amount of data demonstrates that the abundance of Cdt1 is a critical determinant for cell cycle progression. Dysregulation of the Cdt1/CRL4DCAF2 pathway causes re-replication and cellular DNA damage response that includes apoptosis. Cdt1 has become a significant biomarker that mediates the genotoxic effects of anticancer drugs such as MLN4924/Pevonedistat [21] and recently identified CRL4 inhibitor 33-11 [82]. However, the cytotoxicity of MLN4924/Pevonedistat and 33-11 can be rescued only partially by the depletion of Cdt1 [21,82], suggesting contributions by additional factors. Broader investigation of substrate spectrum and biological signaling pathways affected by CRL4 inhibition may lead to identification of previously unknown mechanisms in cancer biology and therapeutics.
Acknowledgement
This work was supported by NIH grant 1R01CA251425-01 to Z-Q P and RJD. We thank N. Zheng of University of Washington, Seattle, for providing the image of E3 CRL4SV5-V used for Figures 1 and 4.
Competing Interest Statement
R.J.D., Z.-Q.P., and K.W. are inventors on patent application 63/144,358 submitted by Icahn School of Medicine at Mount Sinai claiming inhibitors of Cullin- RING E3 ubiquitin ligase 4 (CRL4) to treat leukemia and other cancers.
References
2. Sarikas A, Hartmann T, Pan ZQ. The cullin protein family. Genome Biol. 2011;12(4):220.
3. Li T, Chen X, Garbutt KC, Zhou P, Zheng N. Structure of DDB1 in complex with a paramyxovirus V protein: viral hijack of a propeller cluster in ubiquitin ligase. Cell. 2006;124(1):105-17.
4. Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature. 2006;443(7111):590-3.
5. Rusnac DV, Zheng N. Structural Biology of CRL Ubiquitin Ligases. Adv Exp Med Biol. 2020;1217:9-31.
6. Yamoah Y, Oashi T, Sarikas A, Gazdoiu S, Osman R, Pan ZQ. Auto-inhibitory regulation of SCF-mediated ubiquitination by human cullin 1’s C-terminal tail. PNAS. 2008;105(34):12230-5.
7. Pan ZQ, Kentsis A, Dias DC, Yamoah K, Wu K. Nedd8 on cullin: building an expressway to protein destruction. Oncogene. 2004;23:1985-97.
8. Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell. 2008;134:995-1006.
9. Higa LA, Mihaylov IS, Banks DP, Zheng J, Zhang H. Radiation-mediated proteolysis of Cdt1 by CUL4–ROC1 and CSN complexes constitutes a new checkpoint. Nature Cell Biol. 2003;5:1008-15.
10. Hu J, McCall CM, Ohta T, Xiong Y. Targeted ubiquitination of CDT1 by the DDB1–CUL4A–ROC1 ligase in response to DNA damage. Nature Cell Biol.2004;6:1003-9.
11. Jin J, Arias EE, Chen J, Harper JW, Walter JC. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell. 2006;23(5):709-21.
12. Maiorano D, Moreau J, Mechali M. XCDT1 is required for the assembly of pre-replicative complexes in Xenopus laevis. Nature. 2000;404(6778):622-5.
13. Nishitani H, Lygerou Z, Nishimoto T, Nurse P. The Cdt1 protein is required to license DNA for replication in fission yeast. Nature. 2000;404(6778):625-628.
14. Zhong W, Feng H, Santiago FE, Kipreos ET. CUL-4 ubiquitin ligase maintains genome stability by restraining DNA-replication licensing. Nature. 2003;423( 6942):885-9.
15. Kim Y, Kipreos ET. Cdt1 degradation to prevent DNA re-replication: conserved and non-conserved pathways. Cell Div. 2007;2:18-27.
16. Arias EE, Walter JC. PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent rereplication. Nat Cell Biol. 2006;8(1):84-90.
17. Higa LA, Wu M, Ye T, Kobayashi R, Sun H, Zhang H. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat Cell Biol. 2006;8(11):1277-83.
18. Nishitani H, Sugimoto N, Roukos V, Nakanishi Y, Saijo M, Obuse C, et al. Two E3 ubiquitin ligases, SCFSkp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO J. 2006;25(5):1126-36.
19. Senga T, Sivaprasad U, Zhu W, Park JH, Arias EE, Walter JC, et al. PCNA is a cofactor for Cdt1 degradation by CUL4/DDB1-mediated N-terminal ubiquitination. J Biol Chem. 2006;281(10):6246-52.
20. Xu Y-W, Cao L-R, Wang M, Xu Y, Wu X, Liu J, et al. Maternal DCAF2 is crucial for maintenance of genome stability during the first cell cycle in mice. J Cell Sci. 2017;130(19):3297-07.
21. Pan W-W, Zhou J-J, Yu C, Xu Y, Guo L-J, Zhang H-Y, et al. Ubiquitin E3 ligase CRL4(CDT2/DCAF2) as a potential chemotherapeutic target for ovarian surface epithelial cancer. J Biol Chem. 2013;288(41):29680-91
22. Liontos M, Koutsami M, Sideridou M, Evangelou K, Kletsas D, Levy B, et al. Deregulated overexpression of hCdt1 and hCdc6 promotes malignant behavior. Cancer Res. 2007;67(22):10899-909.
23. Waning DL, Li B, Jia N, Naaldijk Y, Goebel WS, HogenEsch H, et al. Cul4A is required for hematopoietic cell viability and its deficiency leads to apoptosis. Blood. 2008;112(2):320-9.
24. Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res. 2010;70(24):10310-20.
25. Sansam CL, Shepard JL, Lai K, Ianari A, Danielian PS, Amsterdam A, et al. DTL/CDT2 is essential for both CDT1 regulation and the early G2/M checkpoint. Genes Dev. 2006;20(22):3117-29.
26. Zhou P, Yan F. CRL4 Ubiquitin Pathway and DNA Damage Response. Adv Exp Med Biol. 2020;1217:225-39.
27. Abbas T, Sivaprasad U, Terai K, Amador V, Pagano M, Dutta A. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 2008;22(18):2496-2506.
28. Kim Y, Starostina NG, Kipreos ET. The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 2008;22(18):2507-19.
29. Nishitani H, Shiomi Y, Iida H, Michishita M, Takami T, Tsurimoto T. CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J Biol Chem. 2008;283(43):29045-52.
30. Centore RC, Havens CG, Manning AL, Li JM, Flynn RL, Tse A, et al. CRL4 (Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol Cell. 2010;40(1):22- 33.
31. Jorgensen S, Eskildsen M, Fugger K, Hansen L, Larsen MS, Kousholt AN, et al. SET8 is degraded via PCNA-coupled CRL4(CDT2) ubiquitylation in S phase and after UV irradiation. J Cell Biol. 2011;192(1):43-54.
32. Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327(5971):1345-50.
33. Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512(7512):49-53.
34. Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301-5.
35. Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343(6168):305-9.
36. Krönke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature. 2015;523(7559):183-8.
37. Petzold G, Fischer ES, Thomä NH. Structural basis of lenalidomide-induced CK1α degradation by the CRL4(CRBN) ubiquitin ligase. Nature. 2016;532(7597):127-30.
38. Matyskiela ME, Lu G, Ito T, Pagarigan B, Lu CC, Miller K, et al. A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature. 2016;535(7611):252-7.
39. Verma R, Mohl D, Deshaies RJ. Harnessing the Power of Proteolysis for Targeted Protein Inactivation. Mol Cell. 2020;77(3):446-60.
40. Lu G, Weng S, Matyskiela M, Zheng X, Fang W, Wood S, et al. UBE2G1 governs the destruction of cereblon neomorphic substrates. Elife. 2018;7:e40958.
41. Han T, Goralski M, Gaskill N, Capota E, Kim J, Ting TC, et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science. 2017;28:356(6336).
42. Zou Y, Mi J, Cui J, Lu D, Zhang X, Guo C, et al. Characterization of nuclear localization signal in the N terminus of CUL4B and its essential role in cyclin E degradation and cell cycle progression. J Biol Chem. 2009;284:33320-32.
43. Liu L, Lee S, Zhang J, Peters SB, Hannah J, Zhang Y, et al. CUL4A abrogation augments DNA damage response and protection against skin carcinogenesis. Mol Cell. 2009;34(4):451-60.
44. Liu L, Yin Y, Li Y, Prevedel L, Lacy EH, Ma L, et al. Essential role of the CUL4B ubiquitin ligase in extra-embryonic tissue development during mouse embryogenesis. Cell Res. 2012;22(8):1258-69.
45. Chen C-Y, Tsai M-S, Lin C-Y, Yu IS, Chen Y-T, Lin S-R, et al. Rescue of the genetically engineered Cul4b mutant mouse as a potential model for human X-linked mental retardation. Hum Mol Genet. 2012;21(19):4270-85.
46. Cang Y, Zhang J, Nicholas SA, Bastien J, Li B, Zhou P, et al. Deletion of DDB1 in mouse brain and lens leads to p53-dependent elimination of proliferating cells. Cell. 2006;127(5):929-40.
47. Cang Y, Zhang J, Nicholas SA, Kim AL, Zhou P, Goff SP. DDB1 is essential for genomic stability in developing epidermis. Proc Natl Acad Sci USA. 2007;104(8):2733-7.
48. Zhou Z, Song X, Wavelet CM, Wan Y. Cullin 4-DCAF Proteins in Tumorigenesis. Adv Exp Med Biol. 2020.1217:241-59.
49. Wang Y, Zhang P, Liu Z, Wang Q, Wen M, Wang Y, et al. CUL4A overexpression enhances lung tumor growth and sensitizes lung cancer cells to Erlotinib via transcriptional regulation of EGFR. Molecular Cancer. 2014;13:252.
50. Yang Y-L, Hung M-S, Wang Y, Ni J, Mao J-H, Hsieh D, et al. Lung tumourigenesis in a conditional Cul4A transgenic mouse model. Journal of Pathology. 2014;233(2):113-23.
51. Wang Y, Wen M, Kwon Y, Xu Y, Liu Y, Zhang P, et al. CUL4A Induces Epithelial-Mesenchymal Transition and Promotes Cancer Metastasis by Regulating ZEB1 Expression. Cancer Research. 2014;74(2):520-31.
52. Song B, Zhan H, Bian Q, Li J. Knockdown of CUL4B inhibits proliferation and promotes apoptosis of colorectal cancer cells through suppressing the Wnt/beta-catenin signaling pathway. International Journal of Clinical and Experimental Pathology. 2015;8(9):10394-402.
53. Sui X, Zhou H, Zhu L, Wang D, Fan S, Zhao W. CUL4A promotes proliferation and metastasis of colorectal cancer cells by regulating H3K4 trimethylation in epithelialmesenchymal transition. Oncotargets and Therapy. 2017;10:735-43.
54. Yuan J, Han B, Hu H, Qian Y, Liu Z, Wei Z, et al. CUL4B activates Wnt/β-catenin signalling in hepatocellular carcinoma by repressing Wnt antagonists. J Pathol. 2015;235(5):784-95.
55. Yuan J, Jiang B, Zhang A, Qian Y, Tan H, Gao J, et al. Accelerated hepatocellular carcinoma development in CUL4B transgenic mice. Oncotarget. 2015;6(17):15209-21.
56. Chen LC, Manjeshwar S, Lu Y, Moore D, Ljung BM, Kuo WL, et al. The human homologue for the Caenorhabditis elegans cul-4 gene is amplified and overexpressed in primary breast cancers. Cancer Research. 1998;58(16):3677-83.
57. Birner P, Schoppmann A, Schindl M, Dinhof C, Jesch B, Berghoff AS, et al. Human homologue for Caenorhabditis elegans CUL-4 protein overexpression is associated with malignant potential of epithelial ovarian tumours and poor outcome in carcinoma. Journal of Clinical Pathology. 2012;65(6):507-11.
58. Deng J, Lei W, Xiang X, Zhang L, Lei J, Gong Y, et al. Cullin 4A (CUL4A), a direct target of miR-9 and miR- 137, promotes gastric cancer proliferation and invasion by regulating the Hippo signaling pathway. Oncotarget. 2016;7(9):10037-50.
59. Gong Y, Xiang X-J, Feng M, Chen J, Fang Z-L, Xiong J-P. CUL4A promotes cell invasion in gastric cancer by activating the NF-kappa B signaling pathway. Biologics- Targets & Therapy. 2017;11:45-53.
60. Jiang T, Tang H-m, Wu Z-h, Chen J, Lu S, Zhou C-z, et al. Cullin 4B is a novel prognostic marker that correlates with colon cancer progression and pathogenesis. Medical Oncology. 2013;30(2).
61. Zhang J-Q, Chen S, Gu J-N, Zhu Y, Zhan Q, Cheng D-F, et al. MicroRNA-300 promotes apoptosis and inhibits proliferation, migration, invasion and epithelialmesenchymal transition via the Wnt/-catenin signaling pathway by targeting CUL4B in pancreatic cancer cells. Journal of Cellular Biochemistry. 2018. 119(1):1027-40.
62. Jia L, Yan F, Cao W, Chen Z, Zheng H, Li H, et al. Dysregulation of CUL4A and CUL4B Ubiquitin Ligases in Lung Cancer. Journal of Biological Chemistry. 2017;292(7):2966-78.
63. Huang G-K, Liu T-T, Weng S-W, You H-L, Wei Y-C, Chen C-H, et al. (2017) CUL4A overexpression as an independent adverse prognosticator in intrahepatic cholangiocarcinoma. BMC Cancer. 2017;17(1):395.
64. Li P, Zhang L, Yang M, Qi M, Jin X, Han B. Cul4B is a novel prognostic marker in cholangiocarcinoma. Oncology Letters. 2017;14(2):1265-74.
65. Zhang T-J, Xue D, Zhang C-D, Zhang Z-D, Liu Q-R, Wang J-Q. Cullin 4A is associated with epithelial to mesenchymal transition and poor prognosis in perihilar cholangiocarcinoma. World Journal of Gastroenterology. 2017;23(13):2318-29.
66. Liu C-L, Yu I-S, Pan H-W, Lin S-W, Hsu H-C, L2dtl is essential for cell survival and nuclear division in early mouse embryonic development. J Biol Chem. 2007;282:1109-18.
67. Benamar M, Guessous F, Du K, Corbett P, Obeid J, Gioeli D, et al. Inactivation of the CRL4-CDT2-SET8/ p21 ubiquitylation and degradation axis underlies the therapeutic efficacy of pevonedistat in melanoma. EBioMedicine. 2016;10:85-100.
68. Mackintosh C, Ordonez JL, Garcia-Dominguez DJ, Sevillano V, Llombart-Bosch A, Szuhai K, et al. 1q gain and CDT2 overexpression underlie an aggressive and highly proliferative form of Ewing sarcoma. Oncogene. 2012;31(10):1287-98.
69. Vanderdys V, Allak A, Guessous F, Benamar M, Read PW, Jameson MJ, et al. The neddylation inhibitor pevonedistat (MLN4924) suppresses and radiosensitizes head and neck squamous carcinoma cells and tumors. Mol Cancer Ther. 2018;17(2):368-380.
70. Olivero M, Dettori D, Arena S, Zecchin D, Lantelme E, Di Renzo MF. The stress phenotype makes cancer cells addicted to CDT2, a substrate receptor of the CRL4 ubiquitin ligase. Oncotarget. 2014;5(15):5992-6002.
71. Wertz IE, Wang X. From discovery to bedside: Targeting the ubiquitin system. Cell Chem Biol. 2019;26:156-77.
72. Pozhidaeva, A., Valles, G., Wang, F., Wu, J., Sterner, D.E., Nguyen, P., et al. USP7-specific inhibitors target and modify the enzyme’s active site via distinct chemical mechanisms. Cell Chem. Biol. 2017;24: 1501-1512.e5.
73. Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer Nature. 2009;458(7239):732-6.
74. Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ, et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell. 2010;37(1):102-11.
75. Bonvini P, Zorzi E, Basso G, Rosolen A. Bortezomibmediated 26S proteasome inhibition causes cell-cycle arrest and induces apoptosis in CD-30+ anaplastic large cell lymphoma. Leukemia. 2007;21(4):838-42.
76. Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274(5289):948-53.
77. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844-8.
78. Estrada-Ortiz N, Neochoritis CG, Dömling A. How to design a successful p53-MDM2/X interaction inhibitor: a thorough overview based on crystal structures. ChemMedChem. 2016;11(8):757-72.
79. Plechanovová A, Jaffray EG, Tatham MH, Naismith JH, Hay RT. Structure of a RING E3 ligase and ubiquitinloaded E2 primed for catalysis. Nature. 2012;489:115-20.
80. Wu K, Chong RA, Yu Q, Bai J, Spratt DE, Ching K, et al. Suramin inhibits cullin-RING E3 ubiquitin ligases. Proc Natl Acad Sci USA 2016;113:E2011-E2018.
81. Wu K, Pan ZQ. A Novel Strategy to Track Lysine-48 Ubiquitination by Fluorescence Resonance Energy Transfer. Methods Mol Biol. 2021;2267:91-102.
82. Wu K, Huynh KQ, Lu I, Moustakim M, Miao H, Yu C, et al. Inhibitors of cullin-RING E3 ubiquitin ligase 4 with antitumor potential. Proc Natl Acad Sci USA. 2021;118(8):e2007328118.
83. Wu K, Ching K, Chong RA, Pan ZQ. A new FRET-based platform to track substrate ubiquitination by fluorescence. J Biol Chem. 2021;296:100230.
84. Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, et al. The promise and peril of chemical probes. Nat Chem Biol. 2015;11:536-41.
85. Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703-9.
86. Cardote TAF, Gadd MS, Ciulli A, Crystal structure of the Cul2-Rbx1-EloBC-VHL ubiquitin ligase complex. Structure. 2017;25:901-911.e3.
87. Orlando E, Aebersold DM, Medová M, Zimmer Y, Oncogene addiction as a foundation of targeted cancer therapy: The paradigm of the MET receptor tyrosine kinase. Cancer Lett. 2019;443:189-202.