Abstract
Sarcoidosis is a systemic granulomatous disease that primarily affects adults, but it can also occur in the pediatric population, although less commonly. Common manifestations of sarcoidosis include pulmonary involvement, lymphadenopathy, and skin lesions. The clinical presentation in children can be diverse, making diagnosis challenging. Age-related variations in clinical features have been observed, with distinct manifestations in younger versus older children. We present a case of an 11-year-old boy who exhibited generalized maculopapular skin lesions and lymphadenopathy, which is atypical for pediatric sarcoidosis. The patient had a history of juvenile idiopathic arthritis (JIA). Laboratory evaluation revealed elevated serum angiotensin-converting enzyme (ACE), Histopathological examination of an excised lymph node confirmed the diagnosis. Treatment led to substantial improvement and resolution of symptoms. This case emphasizes the importance of considering sarcoidosis in the differential diagnosis of children presenting with skin lesions and lymphadenopathy. The diverse clinical manifestations of pediatric sarcoidosis underscore the need for further research to establish standardized management strategies for this population. Regular follow-up evaluations are necessary to monitor disease activity and manage potential long-term sequelae.
Keywords
Childhood sarcoidosis, Skin lesions, Lymphadenopathy, Juvenile idiopathic arthritis, Angiotensin-Converting Enzyme
Key Clinical Message
Pediatric sarcoidosis can present with atypical symptoms, including skin lesions and lymphadenopathy, and requires a comprehensive diagnostic approach. Early recognition and multidisciplinary management are crucial for optimizing outcomes and preventing complications in children with sarcoidosis. Clinicians should consider sarcoidosis in the differential diagnosis of children presenting with unexplained skin lesions and lymphadenopathy, particularly in those with a history of juvenile idiopathic arthritis or other autoimmune disorders.
Introduction
Sarcoidosis is a complex granulomatous disease that primarily affects adults, with its true incidence and prevalence in children remaining largely unknown [1]. While the condition is significantly less common in the pediatric population, several larger reviews have documented an incidence of clinically recognized sarcoidosis in children ranging from 0.22 to 0.29 cases per 100,000 children per year. This incidence appears to increase gradually with age, peaking in the teenage years, particularly between 13 and 15 years of age [2]. In the pediatric population, two distinct forms of sarcoidosis are often recognized. Older children and young adults typically present with a multisystemic disease characterized by a combination of lymphadenopathy, pulmonary involvement, ocular manifestations, and cutaneous symptoms, such as erythema nodosum [2,3]. Understanding these patterns is crucial for timely diagnosis and effective management of sarcoidosis in children, as early intervention can significantly impact outcomes and quality of life. We present a rare case of sarcoidosis in an 11-year-old boy who exhibited generalized maculopapular skin lesions and lymphadenopathy, atypical findings in pediatric sarcoidosis. The patient also had a history of juvenile idiopathic arthritis (JIA), which adds complexity to the case. This case is significant due to its unusual clinical presentation and the involvement of specific organs, emphasizing the need for heightened awareness of sarcoidosis in the differential diagnosis of children with similar symptoms. Early recognition and management are crucial for optimizing outcomes and minimizing potential long-term sequelae.
Case History/ Examination
An 11-year-old boy from Afghanistan, currently residing in Iran, presented to the Hematology and Oncology Clinic at Mofid Children's Hospital in June 2023 with complaints of disseminated skin lesions and swollen lymph nodes. The patient was born to unrelated parents through normal term delivery. Upon initial evaluation, a chest X-ray and initial laboratory tests were requested, and the patient was scheduled for a lymph node biopsy. Due to the history of methotrexate use, it was recommended to consult with a rheumatologist before the procedure. Consequently, the patient was referred to the Rheumatology Department.
In the patient’s history; approximately six months ago, the patient developed generalized skin lesions in the form of maculopapular rashes on the face, trunk, and extremities. Two months prior to presentation, he also experienced swelling of the lymph nodes in the axillary, inguinal, clavicular, and submandibular regions. On physical examination, the patient exhibited a blood pressure (BP) of 110/70 mmHg, pulse rate (PR) of 78 beats per minute, respiratory rate (RR) of 16 breaths per minute, and oxygen saturation of 98% on room air. On initial examination, generalized maculopapular rashes were observed on the trunk, abdomen, and extremities. Psoriasiform lesions were noted on the knees, characterized by erythematous, well-demarcated plaques with scaly surfaces, resembling typical findings of psoriasis. Extensive generalized lymphadenopathy was also present, particularly in the axillary and inguinal regions (Figure 1). Generalized, non-tender, mobile lymphadenopathy was present; the largest inguinal nodes measured approximately 3–4 cm (by palpation). Symptoms were associated with reduced participation in physical activity and occasional school absences, with transient social self-consciousness due to visible skin lesions. Additionally, arthritis of the wrist and ankle joints was evident, along with conjunctivitis. A more detailed medical history revealed that the patient had been experiencing arthritis of the wrist, knee, and ankle joints, as well as morning stiffness since the age of six. In Afghanistan, at that time, he had been diagnosed with juvenile idiopathic arthritis and had received treatment with corticosteroids and methotrexate for six months. However, the patient discontinued the medications and did not have proper follow-up. He also mentioned recent weight loss over the past six months. The symptoms and investigations timeline are summarized in Table 1.
|
Date/Day |
Event |
Finding/Action |
Outcome |
|
5 years prior (at age 6) |
JIA symptoms |
Wrist/knee/ankle arthritis; morning stiffness |
Prior corticosteroids + methotrexate for ~6 months (later discontinued) |
|
6 months prior (December 2022) |
Symptom onset (skin) |
Generalized maculopapular rash on face, trunk, and extremities |
— |
|
2 months prior (April 2023) |
Lymphadenopathy onset/progression |
Axillary, inguinal, clavicular, and submandibular regions |
— |
|
Day 0 (June 2023) |
Clinic visit at Mofid Children’s Hospital |
Chest X-ray and baseline labs ordered; Rheumatology consult |
Further evaluation planned |
|
Day 7 |
Physical examination |
Generalized lymphadenopathy; largest inguinal nodes (3–4 cm); psoriasiform plaques on knees; synovitis (wrists/ankles); conjunctivitis |
Baseline status recorded |
|
Day 10 |
Laboratory result |
Elevated serum ACE |
Supports sarcoidosis diagnosis |
|
Day 15 |
Imaging / additional evaluation |
CT: Diffuse ground-glass micronodules (suggestive of ILD); prominent bilateral hilar lymphadenopathy; multiple bilateral axillary lymphadenopathies |
Imaging consistent with sarcoidosis |
|
Day 20 |
Lymph node biopsy |
Excision and pathology |
Non-caseating granulomas |
|
ACE: Angiotensin-Converting Enzyme; CXR: Chest X-ray; CT: Computed Tomography; ILD: Interstitial Lung Disease |
|||
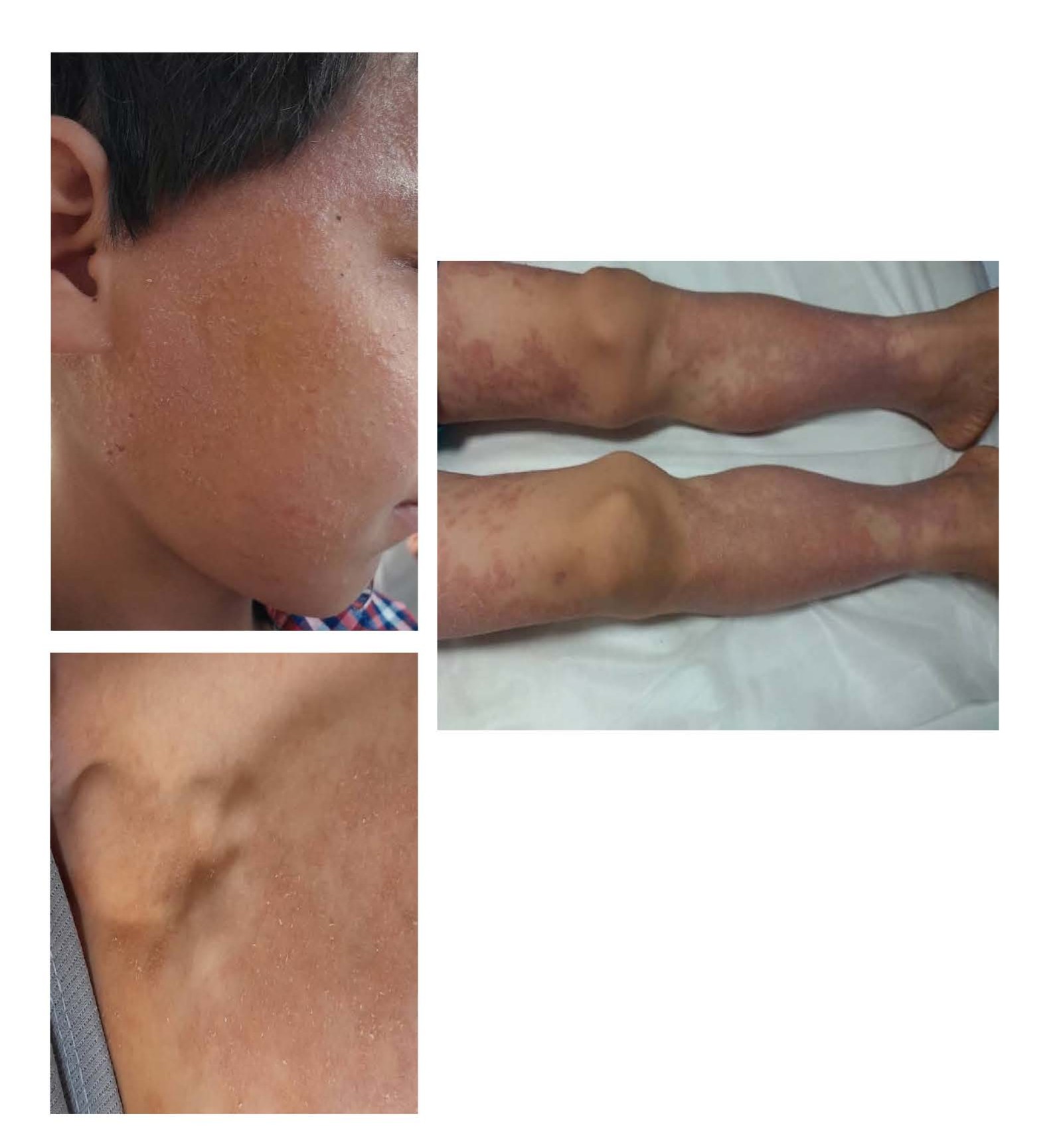
Figure1. The skin manifestations and axillary lymphadenopathies.
Methods (Differential Diagnosis, Investigations and Treatment)
Based on the patient's medical history and clinical findings, lymphoma, sarcoidosis, and NOD2 Blau syndrome were considered in the differential diagnoses. Further initial laboratory tests and imaging were requested, and the patient was hospitalized for a lymph node biopsy. Initial laboratory investigations revealed leukopenia, elevated erythrocyte sedimentation rate (ESR), hypercalcemia, and hypergammaglobulinemia. Rheumatoid factor (RF) and Anti- Cyclic Citrullinated Peptide (CCP) were negative (Table 2). Abdominal and pelvic ultrasound showed multiple portahypatic lymph nodes, with a maximum short-axis diameter of 9 mm, as well as several huge bilateral inguinal lymph nodes (pathologic lymphadenopathy).
|
TEST |
VALUE |
NORMAL RANGE |
TEST |
VALUE |
NORMAL RANGE |
|
WBC (cell/mm3)
|
7,000 |
4,000–12,000 |
ANTI CCP |
NEGETIVE |
NEG<10 |
|
Neutrophil(cell/mm3) |
4,270 |
1,500–8,500 |
ACE |
121 |
NEG<10 |
|
Lymphocyte(cell/mm3)
|
2,240 |
3,000–9,500 |
LDH |
500 |
< 746
|
|
Hb (gr/dl) |
12.8 |
11.5–13.5 |
CPK |
36 |
24–229 |
|
Plt |
326000 |
150,000–450,000 |
Uric Acid |
5.3 |
3.6–4 |
|
ESR (mm/hr.) |
23 |
NL<20 |
IgM (mg/dl) |
65 |
31–179 |
|
CRP (mg/L) |
4 |
NL<6 |
IgG (mg/dl) |
1,020 |
700–1,400 |
|
ANA |
1/1,280 Fine speckled |
NEG<1/40 POS>1/80 |
IgA (mg/dl) |
396 |
40–350 |
|
RF |
NEGATIVE |
NEG<10 |
IgE (IU/ml) |
60 |
Up to 200 |
|
Calcium |
11 |
7/5–9/5 |
AST |
50 |
NL<30 |
|
Ph |
4.6 |
|
ALT |
15 |
NL<30 |
|
U ca/U cr |
2 |
NL<0/5 |
ALK-p |
354 |
NL<250 |
|
PPD TEST |
NEGATIVE |
|
BUN |
14.2 |
6–20 |
|
IGRA |
NEGATIVE |
|
Cr |
0.7 |
0.6–1.4 |
|
WBC: White Blood cell; Hb: Hemoglobin; PLT: Platelet; ANA: Anti-Nuclear Antibody; RF: Rheumatoid Factor; Anti CCP: Anti Cyclic Citrullinated Peptide; ESR: Erythrocyte Sedimentation Rate; CRP: C-reactive Protein; Ig: Immunoglobulin; ACE: Angiotensin-Converting Enzyme; ALT: Alanine Transaminase; AST: Aspartate Transaminase; Alk-p: Alkaline Phosphatase; PPD: Tuberculin Test; IGRA: Interferon-Gamma-Release Assay; BUN: Blood Urine Nitrogen. As mentioned in above table, as a bold values; Our patient has, Elevated acute phase reactants, Hypercalcemia, Elevated ACE |
|||||
A chest X-ray was performed as part of the initial evaluation to assess pulmonary involvement, which is common in sarcoidosis. The X-ray findings indicated potential lung involvement, prompting further investigation. Subsequently, a chest CT scan was indicated to provide a more detailed assessment of the lung parenchyma and lymphatic structures. The CT scan revealed diffuse centrilobular ground-glass micronodules in both lung fields, suggestive of interstitial lung disease associated with sarcoidosis. Additionally, two small nodules (4 mm) were identified in the right middle lobe and the superior segment of the right lower lobe. The bilateral hilar regions were prominently enlarged, indicating lymphadenopathy, which is characteristic of sarcoidosis, along with multiple bilateral axillary lymphadenopathies. These imaging findings were instrumental in confirming the diagnosis and guiding the subsequent management plan. Additionally, the ACE level was found to be elevated. The investigations for tuberculosis were negative. Although the patient’s cutaneous manifestations were prominent, we prioritized excisional lymph-node biopsy to secure a single, high-yield tissue diagnosis. The skin lesions were non-ulcerative papules/plaques located on cosmetically sensitive areas; In the context of elevated serum ACE, hypercalcemia, and characteristic chest CT findings (diffuse centrilobular ground-glass micronodules, bilateral hilar lymphadenopathy, and small right-sided nodules), deferring a skin biopsy minimized procedural burden and potential scarring in a pediatric patient. Consequently, the patient underwent excisional biopsy of a lymph node, and the pathology report confirmed sarcoidosis (non-necrotizing granulomatous inflammation). In our differential diagnosis, we considered lymphoma, but it was excluded due to the absence of B-symptoms (fever, night sweats, weight loss) and the lack of characteristic lymph node morphology on imaging (the absence of necrosis or marked extranodal involvement). Blau syndrome was also considered, but this was ruled out based on the patient’s age of onset, absence of granulomatous inflammation in the skin, and the presence of non-caseating granulomas on biopsy, which are more characteristic of sarcoidosis.
The patient’s ANA titer was 1:1280. In pediatric populations—particularly those with a history of juvenile idiopathic arthritis (JIA)—ANA positivity can occur and is nonspecific. In this case, there were no clinical features suggestive of systemic lupus erythematosus or an overlap connective-tissue disease at presentation, and the lymph-node histopathology confirming non-caseating granulomas established the diagnosis of sarcoidosis. We therefore interpret the elevated ANA as a background autoimmune marker in the setting of prior JIA rather than evidence of an alternative primary diagnosis. The patient is being followed longitudinally for potential autoimmune sequelae.
Treatment and follow-up
Following the histopathologic diagnosis of sarcoidosis, the patient received high-dose pulse methylprednisolone (30 mg/kg/dose; max 1 g/day for 3 days) for acute inflammation control. This was followed by oral prednisolone 2 mg/kg/day with a structured 12-week taper adjusted to clinical and laboratory response: weeks 1–2 at 2 mg/kg/day; weeks 3–4 at ~1.5 mg/kg/day; weeks 5–6 at ~1 mg/kg/day; weeks 7–8 at ~0.5 mg/kg/day; weeks 9–10 at ~0.25 mg/kg/day; weeks 11–12 at ~0.1–0.2 mg/kg/day, then continued at the lowest effective dose with planned taper to cessation as clinically appropriate. Methotrexate (MTX) 10 mg/m² once weekly with folic acid 1 mg daily supplementation was initiated as a steroid-sparing agent and has been continued through the 11-month follow-up based on sustained clinical benefit and tolerability. Safety labs (CBC, AST/ALT, creatinine) were obtained at baseline and every 4–8 weeks, with no dose-limiting adverse effects observed. Re-evaluation for taper or discontinuation is planned at 12 months contingent on disease control. Regular monitoring for potential side effects was established throughout this period. In addition to these treatments, the patient presented with hypercalcemia, a known complication of sarcoidosis. To address this, we implemented dietary modifications to reduce calcium intake and initiated hydration therapy with intravenous fluids to promote renal excretion of calcium. Serum calcium levels were monitored closely, and corticosteroids were administered, as they can help reduce hypercalcemia by decreasing granuloma formation and calcium mobilization.
Results (Outcome and Follow-up)
The patient was reviewed every 2–4 weeks during the steroid taper and every 8–12 weeks thereafter. Each visit included targeted examination (skin/lymph nodes, joints, growth/vitals) and laboratory surveillance (CBC, liver enzymes, creatinine, calcium); ACE was trended as an adjunct marker of disease activity. Given ocular involvement, we obtained a baseline slit-lamp examination, which showed bilateral posterior iris synechiae and inactive uveitis; no topical anti-inflammatory therapy was required, and re-evaluation at 3–6 months was planned. Over 11 months of follow-up, the patient demonstrated sustained clinical improvement, with complete resolution of cutaneous lesions and marked regression of lymphadenopathy (Figure 2). Energy levels and school attendance returned to baseline, and no functional limitations were observed during follow-up. The patient continues methotrexate with scheduled surveillance as outlined and regular follow-up appointments are scheduled to monitor his condition. The treatment and follow-up timeline are summarized in Table 3.
|
Date/Day |
Event |
Finding/Action |
Outcome |
|
Day 0 |
Treatment initiated |
Pulse methylprednisolone 30 mg/kg/dose (max 1 g) × 3 days |
Acute inflammation controlled |
|
Weeks 1–12 |
Steroid taper |
Prednisolone taper per schedule (2 → 1.5 → 1 → 0.5 → 0.25 mg/kg/day →0.1–0.2 mg/kg/day) |
Progressive improvement; no complications |
|
Day 4 |
Steroid-sparing therapy |
Methotrexate 10 mg/m² weekly + folic acid; planned ≥6 months |
Maintains control; reduces steroid exposure |
|
Every 8–12 weeks |
Safety monitoring |
CBC, AST/ALT, creatinine; calcium; ACE trend |
Within acceptable ranges / improved |
|
Month 11 |
Follow-up |
Exam ± imaging; ophthalmology per schedule |
Sustained improvement; rash resolved; lymphadenopathy decreased |
|
CBC: Complete Blood Count; ALT: Alanine Transaminase; AST: Aspartate Transaminase; ACE: Angiotensin-Converting Enzyme |
|||
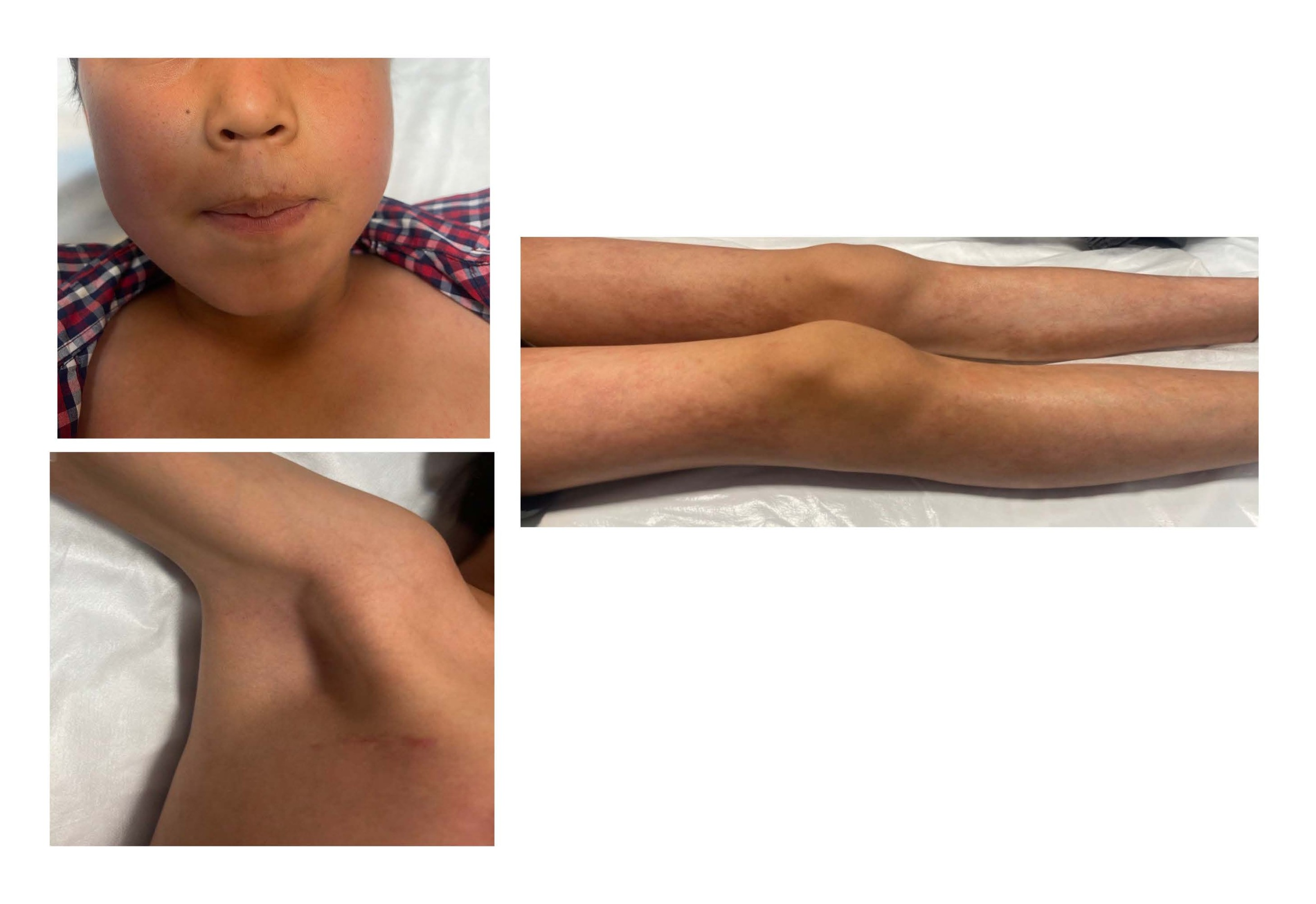
Figure 2. After 2-month follow-up and improvement of skin lesions and lymphadenopathy.
This approach underscores the importance of clinical evaluation and monitoring in managing skin manifestations, illustrating that in certain cases, invasive procedures like biopsy may be unnecessary when the clinical context and treatment response aligns with the diagnosis.
Discussion
Sarcoidosis is a systemic granulomatous disease that primarily affects adults, with relatively rare pediatric cases [1]. True prevalence in children is uncertain because many presentations are subclinical and may resolve spontaneously without diagnosis [2,3]. Pediatric sarcoidosis is heterogeneous and therefore diagnostically challenging [4]. However, it is important to consider sarcoidosis as a potential diagnosis in children presenting with unusual manifestations, as demonstrated in this case of an 11-year-old boy. Age-related patterns are recognized: early-onset (<4 years) disease often presents with the triad of rash, arthritis, and uveitis, whereas school-age and adolescent patients more commonly exhibit pulmonary, lymph-node, and ocular involvement [2,4]. Our 11-year-old patient had generalized maculopapular eruptions and generalized lymphadenopathy with ocular findings—features that align with the older-child pattern, albeit with unusually prominent cutaneous disease, which are atypical manifestations of sarcoidosis in children. Skin involvement in sarcoidosis can manifest in various forms, including erythema nodosum, maculopapular eruptions, plaques, or lupus pernio-like lesions [5]. Lymphadenopathy is a common finding, and it can involve different regions such as the axillary, inguinal, and submandibular lymph nodes. Other commonly affected organ systems include the lungs, eyes, liver, and spleen [2,4]. As S. Kathleen Clark M.D (1987) reported, childhood sarcoidosis cases often occur between the ages of 9 to 15 years, with occasional clusters of cases observed in children under the age of 4 [6]. Recognizing these age-linked phenotypes can shorten time to diagnosis and guide a targeted work-up in children with atypical dermatologic presentations [6]. Hilar lymph node enlargement is a common finding in childhood sarcoidosis, as observed in this case. However, Edwin L. Kendig Jr. noted that hilar lymph node enlargement was practically always present [7]. Ocular lesions are also frequently present, and serious ocular manifestations are not uncommon [4,7]. Therefore, it is crucial to assess and monitor ocular health in children with sarcoidosis. Laboratory findings such as hyperglobulinemia, eosinophilia, leukopenia, hypercalcemia, hypercalciuria, and elevated alkaline phosphatase can provide diagnostic clues and aid in the evaluation of suspected cases [7,8]. The diagnosis of sarcoidosis is based on clinical presentation, imaging studies, histopathological examination, and exclusion of other potential causes [1,9]. Laboratory investigations often show nonspecific findings, but an elevated serum angiotensin-converting enzyme (ACE) level can be supportive [10]. In more than 50% of children with late-onset sarcoidosis, there is an elevation in the serum level of ACE [8]. However, it is important to note that this test is not specific for sarcoidosis, as increased serum ACE activity can also be observed in various other disorders [8,11]. It is worth mentioning that serum levels of ACE (angiotensin-converting enzyme) are elevated in various granulomatous diseases, including sarcoidosis, silicosis, and military tuberculosis. It has generally been accepted that ACE has particular value in indicating the disease activity of sarcoidosis [12]. Therefore, other potential causes of granuloma should be considered and excluded [2,12]. Imaging studies, such as chest X-ray and CT scan, can reveal characteristic findings of sarcoidosis, including bilateral hilar lymphadenopathy and lung parenchymal involvement [1,10]. Histopathological examination of the excised lymph node confirmed the diagnosis of sarcoidosis in this case, demonstrating non-necrotizing granulomatous inflammation. Although genetic consultation was recommended due to possible NOD2 involvement, it was not performed because of cost constraints. Variants in NOD2 are implicated in granulomatous disorders such as Blau syndrome/early-onset sarcoidosis, which can present with arthritis, uveitis, and dermatitis. While we could not perform genetic testing, NOD2-related mechanisms remain biologically plausible in pediatric granulomatous disease. Future access to testing could help clarify whether an underlying monogenic pathway contributes to this presentation. Prior studies also associate certain NOD2 variants with sarcoidosis susceptibility, particularly in adults of European descent, underscoring the rationale for genetic evaluation when feasible [13]. It should be noted that our patient had a history of juvenile idiopathic arthritis (JIA) and had previously been treated with corticosteroids and methotrexate. The literature has described an association between sarcoidosis and juvenile idiopathic arthritis (JIA), suggesting a possible link between these two conditions. UKAE S, et al, reported nine cases of sarcoidosis in Japanese children aged 4 years or younger who were initially misdiagnosed with JIA [14]. Similarly, Rosenberg AM reported a case of a 6-year-old girl with arthropathy accompanying sarcoidosis [15]. In addition, Mallory SB's study presented cases of children who displayed features indicative of both sarcoidosis and juvenile rheumatoid arthritis (JIA), with some cases exhibiting affected family members. In their clinical experience, four children with sarcoidosis and severe joint manifestations were observed, two of whom had a personal or family history of JIA. Notably, three out of the four children also displayed ichthyosiform cutaneous manifestations, suggesting a potential association between severe joint disease and ichthyosiform changes. Therefore; Mallory SB suggests that biopsy of cutaneous lesions should be considered in children presenting with such symptoms [16]. Based on previous studies, early-onset sarcoidosis can often be mistaken for systemic-onset JIA due to their overlapping systemic manifestations such as fever, weight loss, and fatigue. However, distinct skin changes can help differentiate between the two diseases at the onset. The rash associated with JIA is typically pink, transient, and macular, while sarcoidosis is characterized by papules or plaque-like lesions with scaling. Arthritis in sarcoidosis is typically painless with boggy effusions of the synovium and no limitation of range of motion [15,16]. However, cases of painful and destructive polyarthritis resembling JIA have been described in early-onset sarcoidosis [10,16]. In our case, the long-standing arthritis beginning at age six (compatible with JIA), together with biopsy-proven non-caseating granulomas during the current presentation, supports co-morbidity rather than a prior misdiagnosis. We therefore interpret the patient’s JIA history as background autoimmunity co-existing with new-onset sarcoidosis. The high ANA titer (1:1280) is nonspecific in pediatric rheumatology and, in this context, is more consistent with background JIA-related autoimmunity than with an alternative unifying diagnosis. This case illustrates an uncommon but clinically relevant overlap between autoimmune (JIA) and granulomatous (sarcoidosis) processes in childhood, highlighting diagnostic pitfalls and the value of a structured, timeline-driven evaluation. Therefore, it is important to differentiate early-onset sarcoidosis, which predominantly affects children under 8 years of age, from later-onset sarcoidosis that primarily affects older children and young adults. While there are differences in clinical presentation, symptoms, course, and prognosis between the two types, certain clinical features and histological findings can overlap [2,17]. Management of sarcoidosis in children is challenging due to the limited evidence and lack of standardized guidelines [4,10]. Treatment strategies are often extrapolated from the adult population [10]. In this case, the patient was initiated on high-dose pulse methylprednisolone followed by an oral prednisolone tapering regimen. Methotrexate was also prescribed as a steroid-sparing agent. Regular follow-up assessments showed significant improvement, with complete resolution of skin lesions and lymphadenopathy. Mortality in childhood sarcoidosis is estimated to be around 5%, indicating that the disease can have serious consequences. Long-term sequelae are observed in approximately 10% to 20% of cases [6]. Early recognition and diagnosis of childhood sarcoidosis play a crucial role in preventing complications such as blindness, pulmonary insufficiency, and renal impairment [4,10]. Timely intervention and appropriate management can help optimize outcomes and improve the overall prognosis for affected children. The prognosis of pediatric sarcoidosis varies, with some cases exhibiting a self-limiting course while others require long-term management [1,4]. The long-term course and prognosis of childhood sarcoidosis, particularly early-onset disease, are not well established [2]. The disease can have relapses and remissions, necessitating close monitoring and adjustment of treatment [4,10]. Regular follow-up evaluations are crucial to assess disease activity, monitor organ involvement, and manage potential complications [2, 4].
Conclusion
In summary, sarcoidosis is a complex and heterogeneous disease that can affect children, with distinct age-related variations in clinical presentation. This case highlights the importance of considering skin lesions and lymphadenopathy in the differential diagnosis of pediatric sarcoidosis, as they can be atypical manifestations in children. Diagnosis requires a comprehensive approach, including clinical presentation, imaging studies, histopathological examination, and exclusion of other potential causes. The management of pediatric sarcoidosis requires a multidisciplinary approach, but the lack of standardized guidelines for children necessitates extrapolation from adult guidelines. Regular follow-up assessments are crucial for monitoring disease activity and managing complications. Early recognition and appropriate management are essential for optimizing outcomes and improving the prognosis for children with sarcoidosis. Further research is needed to elucidate the long-term course of the disease in children, establish standardized management guidelines, and develop effective treatment strategies tailored to the unique needs of pediatric patients.
Author Contributions
Pooneh Tabibi: Conceptualization; data curation; writing – original draft; writing – review and editing, Vadood Javadi Parvaneh: Project administration, supervision.
Ethical Approval and Consent for Publication
Written informed consent was obtained and signed from the patient and the patient's parents regarding the use of the patient's health information for the purpose of writing and publishing a case report and any accompanying images.
Consent for Publication
The consent for publication was obtained from the patient's parents.
Conflicts of Interest
The authors declare no conflict of interest regarding the publication of this case report.
Acknowledgments
Not applicable.
Funding Information
Not applicable.
Availability of Data and Materials
All data and materials used in this research are available upon request. Researchers interested in accessing the data and materials may contact Email: dr.rheumatology.iran@gmail.com.
References
2. Chiu B, Chan J, Das S, Alshamma Z, Sergi C. Pediatric Sarcoidosis: A Review with Emphasis on Early Onset and High-Risk Sarcoidosis and Diagnostic Challenges. Diagnostics (Basel). 2019 Oct 25;9(4):160.
3. Pattishall EN, Kendig EL Jr. Sarcoidosis in children. Pediatr Pulmonol. 1996 Sep;22(3):195–203.
4. Cimaz R, Ansell BM. Sarcoidosis in the pediatric age. Clin Exp Rheumatol. 2002 Mar-Apr;20(2):231–7.
5. Mañá J, Marcoval J. Skin manifestations of sarcoidosis. Presse Med. 2012 Jun;41(6 Pt 2):e355-74.
6. Clark SK. Sarcoidosis in children. Pediatr Dermatol. 1987 Dec;4(4):291–9.
7. KENDIG EL Jr. Sarcoidosis among children. A review. J Pediatr. 1962 Aug;61:269–78.
8. Hoffmann AL, Milman N, Byg KE. Childhood sarcoidosis in Denmark 1979-1994: incidence, clinical features and laboratory results at presentation in 48 children. Acta Paediatr. 2004 Jan;93(1):30–6.
9. Nathan N, Sileo C, Calender A, Pacheco Y, Rosental PA, Cavalin C, Macchi O, et al. French Sarcoidosis Group (GSF); Silicosis Research Group. Paediatric sarcoidosis. Paediatr Respir Rev. 2019 Feb;29:53–9.
10. Shetty AK, Gedalia A. Childhood sarcoidosis: A rare but fascinating disorder. Pediatr Rheumatol Online J. 2008 Sep 23;6:16.
11. Rodriguez GE, Shin BC, Abernathy RS, Kendig EL Jr. Serum angiotensin-converting enzyme activity in normal children and in those with sarcoidosis. J Pediatr. 1981 Jul;99(1):68–72.
12. Kwon CI, Park PW, Kang H, Kim GI, Cha ST, Kim KS, et al. The usefulness of angiotensin converting enzyme in the differential diagnosis of Crohn's disease and intestinal tuberculosis. Korean J Intern Med. 2007 Mar;22(1):1–7.
13. Anantharajah A. Assessment of clinical, cellular and molecular determinants in sarcoidosis. Master's thesis, The Australian National University, Australia; 2022.
14. Ukae S, Tsutsumi H, Adachi N, Takahashi H, Kato F, Chiba S. Preschool sarcoidosis manifesting as juvenile rheumatoid arthritis: a case report and a review of the literature of Japanese cases. Acta Paediatr Jpn. 1994 Oct;36(5):515–8.
15. Rosenberg AM, Yee EH, MacKenzie JW. Arthritis in childhood sarcoidosis. J Rheumatol. 1983 Dec;10(6):987-90.
16. Mallory SB, Paller AS, Ginsburg BC, McCrossin ID, Abernathy R. Sarcoidosis in children: differentiation from juvenile rheumatoid arthritis. Pediatr Dermatol. 1987 Dec;4(4):313–9.
17. Häfner R, Vogel P. Sarcoidosis of early onset. A challenge for the pediatric rheumatologist. Clin Exp Rheumatol. 1993 Nov-Dec;11(6):685–91.