Abstract
Heat shock proteins (HSPs) are ubiquitous proteins that play an important role in cellular stress responses, contributing to immune activation through interactions with toll-like receptors (TLRs) and other pattern recognition receptors. Despite extensive research, the exact mechanism by which HSPs modulate immune responses is still unclear. This study explores the immune-modulatory role of HSPA1A by examining its effect on cytokine production in PMA-differentiated THP-1 macrophages, preincubated with TLR-specific blocking peptides prior to exposure to recombinant HSPA1A. The results showed that treatment with HSPA1A significantly increased the secretion of both pro-inflammatory cytokines, such as IL-1β and TNF-α, and anti-inflammatory cytokines, such as IL-4 and IL-10, emphasizing its dual role in immune regulation. Blocking peptides targeting TLR2, TLR4, TLR5, and TLR7 were used to study the involvement of these receptors in HSPA1A-induced cytokine production. Further analysis revealed that HSPA1A modulates key signaling pathways, including MyD88, MAPK, and NF-κB. These findings suggest that HSPA1A-TLR interactions and their downstream signaling cascades can be potential therapeutic targets for modulating immune responses in inflammatory and autoimmune diseases.
Keywords
Immune response, Cytokines, THP-1, HSPA1A, TLRs
Introduction
Heat shock proteins (HSPs), also known as stress proteins, are ubiquitous and conserved proteins that have a fundamental role in the molecular chaperone system of cells [1]. HSPs are generally expressed intracellularly [2], however they are overexpressed in response to cellular stress [3]. The overproduction of HSP results in their increased surface expression and release into the extracellular environment, via active secretion or apoptosis [1].
Several studies have indicated that extracellular HSPs, predominantly released endogenously during cellular stress or necrosis, can act as a danger signal [4] and play a role in both innate and adaptive immunity [5]. Certain HSPs can stimulate the production of inflammatory cytokines, chemokines and co-stimulatory molecules through the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway [6]. Furthermore, HSPs have been reported to stimulate cytokine production via the activation of both NF-κB and mitogen-activated protein kinase (MAPK) pathways, which may contribute to the pathogenesis of autoimmune and chronic inflammatory diseases [7]. For example, Moroi et al. [8] showed that extracellular HSP70 can trigger cytokine secretion by activating dendritic cells. HSPA1A is the primary heat- inducible member of the Hsp70 family in humans, with a molecular weight of 72kDa [9]. HSPA1A interacts with target cells through various cell surface receptors, including toll like receptors (TLRs) 2 and 4 [10]. TLRs are a type of pattern recognition receptor playing an important role in recognizing pathogen associated molecular patterns (PAMPs) [11]. Upon binding with a ligand, TLRs recruit toll/interleukin-1 receptor (TIR) domain-containing adaptor proteins, such as myeloid differentiation primary-response protein 88 (MyD88) and TIR domain-containing adaptor inducing IFN-β (TRIF), which initiate signal transduction pathways leading to the activation of NF-κB and MAPK, thereby regulating cytokine production [12]. The main difference in TLR signaling pathways is the recruitment of either MyD88 or TRIF. MyD88 is utilized by all TLRs leading to enhanced expression of proinflammatory cytokine genes, with its activation facilitated by TIRAP or Mal [13].
Primarily released from endogenous sources, HSPA1A enters the extracellular environment through mechanisms such as transport with transmembrane domain proteins, secretion through lysosomal endosomes, or inhibition of phospholipase C activity [14]. Moreover, Saito et al. [15] noted that HSPA1A can also be released passively from damaged or necrotic cells. Extracellular HSPA1A has been shown to influence both pro- and anti-inflammatory responses. It can modulate the immune response by binding to receptors on immune cells, such as natural killer cells and macrophages [5]. This role is significant in cancer biology; for example, Wu et al. [16] observed that extracellular HSP modulates immune response in hepatocarcinoma, thereby contributing to tumor immune evasion. Additionally, a study by Chiricosta et al. [17] reported the upregulation of HSPA1A in multiple sclerosis (MS) and their role in exacerbating immune response in MS [35]. This suggests that intracellular HSPs protect cells under stress, however their overexpression and subsequent release from cells leads to the activation of the immune system [17]. The complex role of HSPs, especially in their interactions with TLRs and the resulting immune responses, highlights the need for a thorough understanding of the precise mechanisms of this interaction as it could be beneficial in treating diseases caused by them [2].
Materials and Methods
THP-1 cell cultures
The human monocytic leukemia cell line, THP-1, was purchased from the American Type Culture Collection (ATCC) and it was cultured in suspension in T75 flasks using RPMI-1640 media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37°C in a humidified tissue culture incubator with 5% CO2. The cells were routinely sub-cultured every 2-3 days, or when confluent and the cell suspension was regularly monitored to ensure optimal growth and tested for viability using the trypan blue exclusion method.
Preparation and differentiation of THP-1 monocytes into macrophages
THP-1 cells with a seeding density of 500,000 cells/ml were centrifuged at 500 g for 5 minutes at 25°C. Following, the supernatant was discarded, and the pellet was resuspended in 10% heat-inactivated RPMI-1640 (HIRPMI-1640) medium. To induce the differentiation of THP-1 monocytic cells into macrophages, phorbol 12-myristate 13-acetate (PMA) was added to the cell suspension at a final concentration of 10 ng/mL. The treated cell suspension was then transferred into 12-well plates with a final volume of 1 ml per well and into 6-well plates with a final volume of 3 ml per well. The culture plates were then incubated at 37°C with 5% CO2 for 24 hours. The progression of cell differentiation was monitored by observing morphological changes under a light microscope. Following differentiation, media containing PMA was carefully discarded, and the cells were gently washed with fresh media to remove any residual PMA. These cells were then subjected to treatment with HIRPMI (control), HSPA1A, TLR-specific blocking peptide and combination of HSPA1A and TLR-specific blocking peptide.
Treatment of THP-1 macrophages with TLR-specific blocking peptides and recombinant HSPA1A
Blocking peptides specific for TLR2, TLR4, TLR5, and TLR7 were prepared by diluting them in 10% HIRPMI-1640 medium. The THP-1 macrophages were then preincubated with these blocking peptides at a final concentration of 100 ng/ml for 30 minutes. Recombinant HSPA1A was purchased from Stressmarq Biosciences, which guarantees the product to be >90% pure and free of endotoxins. The protein was added to THP-1 macrophages at a final concentration of 1000 ng/ml, and the cell cultures were incubated at 37°C with 5% CO2 for 18 hours.
Detection of IL-1β, IL-4, IL-10, and TNF-α concentrations by ELISA
Following treatment, the cell suspension was transferred into a falcon tube and centrifuged twice at 500 g for 5 minutes. The supernatant was then collected for cytokine detection using ELISA, following manufacturer’s protocol (Invitrogen, California, USA). The absorbance was measured at a wavelength of 450 nm using an LT-4500 ELISA plate reader by Labtech. The absorbance values were converted to cytokine concentrations using Prism software.
Detection of MyD88, MAPK, and NF-κB Concentrations by ELISA
After treatment, cells were collected, centrifuged at 1000 RPM for 10 minutes, and the supernatant was discarded. The pellet was dried, resuspended in PBS, and centrifuged at 500 RPM for 3 minutes. After discarding the supernatant, the pellet was dried and loosened. A lysis buffer with 1% (v/v) non-ionic detergent P40 (NP40) with one protease inhibitor tablet in distilled water) was prepared. The pellet was resuspended in 250 ul of lysis buffer and incubated on ice for 30 minutes. Centrifugation at 9000 RPM for 10 minutes yielded the protein fraction, confirmed by Nanodrop.
MyD88 was detected by ELISA according to manufacturer’s instruction (Abcam) and MAPK and NF-κB detected using kit from Invitrogen. The absorbance was measured at 450 nm using an LT-4500 ELISA plate reader, and values were converted to cytokine concentrations using Prism software.
Detection of NF-κB by RT-PCR
The media was removed from the 6-well plates, and cells were washed with PBS. Then 0.1-0.25 % trypsin was added for 5 minutes at room temperature. After detachment 10% RPMI-1640 media was added to inactivate trypsin. The cell suspension was transferred to an RNAse-free falcon tube, centrifuged at 1.6 RPM for 5 minutes and all supernatant was removed. RNA extraction was performed by using RNeasy mini kit by Qiagen, and RNA concentration was confirmed via Nanodrop.
|
Primer |
Sequence |
|
NF-κB forward primer |
5’- FGC GCT TCT CTG CCT TCC TTA -3’ |
|
NF-κB Reverse primer |
5’ - TCT TCA GGT TTG ATG CCC CC -3’ |
|
GAPDH2 Forward primer |
5’ - GAG TCA ACG GAT TTG GTC GT -3’ |
|
GAPDH2 Reverse primer |
5’ - GAC AAG CTT CCC GTT CTC AG -3’ |
For reverse transcription, 12 ul of RNA template was mixed with 1 ul of oligo(dT), incubated at 65°C for 5 minutes using a PCR machine, then placed on ice. Next, 4 ul of reaction buffer, 2 ul of dNTP, and 1 ul reverse transcriptase were added, followed by 60 minutes at 42°C and 5 minutes at 70°C. The samples were diluted in a 1:3 ratio with water. Primers (100 nmol/ul, diluted 1:50) were used for each reaction. Each of the 15 samples had three reactions: no template control, NF-κB primer, and GAPDH primer. RT-PCR cycling conditions: 40 cycles at 95°C for 5 seconds, then 60°C for 30 seconds.
The CT obtained were used to calculate fold change in gene expression using ΔΔCt method.
Fold change= 2 ^−ΔΔCT
Statistical analysis
All different treatments for cytokine detection via ELISA were conducted in triplicate. Data analysis was performed with GraphPad Prism™ version 10.2.3 ((GraphPad Software, Inc, San Diego, CA, USA). A two-way ANOVA was utilized to analyze the differences in time course effects between HSPA1A treatment and the control (treated with 10% HIRPMI only). An ordinary one-way ANOVA was performed to assess the differences among the various treatment groups. For the detection of NF-κB by RT-PCR, where only two biological replicates were available, the Mann-Whitney test was used to evaluate the fold change differences in NF-κB expression between the different treatments.
Results
Figure 1 illustrates the production levels of IL-1β, IL-4, IL-10, and TNF-α cytokines in THP-1 macrophages after treatment with HSPA1A. Cytokine levels were measured at 0 h (pre-treatment) and at 18 h (post-treatment). Notably, there was a significant increase in the secretion of all four cytokines after 18 hours of treatment with HSPA1A compared to the control (10% HIRPMI). Specifically, IL-1β reached 41.77 pg/ml, IL-4 was 42.50 pg/ml, IL-10 was 47.29 pg/ml and TNF-α was 184.6 pg/ml. These increases were statistically significant (p<0.0001), indicating enhanced cytokine production in THP-1 macrophages following HSPA1A treatment.
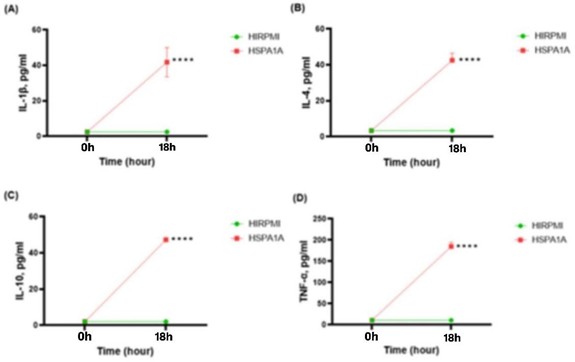
Figure 1: Time course effects of 1000 ng/ml HSPA1A on the secretion of IL-1β, IL-4, IL-10 and TNF-α cytokines from THP-1 macrophages differentiated with PMA. Cytokine levels (pg/ml) were determined at 0 hours and 18 hours post-treatment. The graphs show comparative data for HIRPMI (control) and HSPA1A (treatment). Data are presented as mean ± SD, n = 3 and analyzed using two-way ANOVA with Bonferroni’s multiple comparison post hoc. Significant differences between HSPA1A and HIRPMI treatment are shown **** (p< 0.0001).
The effects of HSPA1A and TLR 2 blocking peptide (TLR2 BP) on the production of inflammatory cytokines were assessed in THP-1 macrophages. As illustrated in Figure 2, HSPA1A significantly increased the secretion of IL-1β (Figure 1A), IL-4 (Figure 1B), IL-10 (Figure 1C), and TNF-α (Figure 1D) compared to the control (HIRPMI) (p<0.0001). When THP-1 macrophages were pre-treated with TLR2 BP, the cytokine levels induced by HSPA1A were significantly reduced (IL-1β: p<0.01, IL-4, IL10 and TNF-α: p<0.0001). This reduction was observed across all cytokines measured, suggesting that blocking TLR2 impacts the effect of HSPA1A on cytokine production [43].
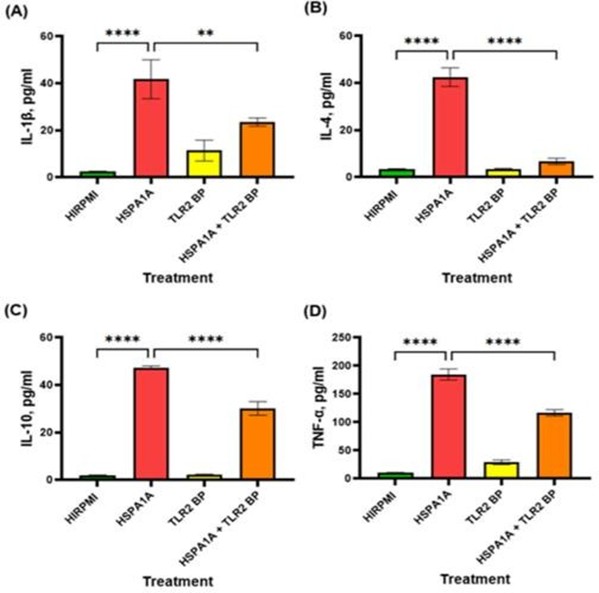
Figure 2: Effects of various treatments on IL-1β, IL-4, IL-10, and TNF-α cytokine production in THP-1 macrophages. The treatments include HIRPMI (control), HSPA1A, TLR2 blocking peptide (TLR2 BP), and a combination of HSPA1A with TLR2 BP. Data are shown as mean concentration (pg/ml) ± SD, n = 3 and tested by Ordinary one-way ANOVA with Bonferroni’s multiple comparison post hoc. Significance levels are indicated as follows: ** (p<0.01) and **** (p<0.0001), comparing treated groups to the control and between specific treatments.
The data presented in Figure 3 illustrates the effects of HSPA1A, TLR4 blocking peptide (TLR4 BP) and their combination on cytokine production. In panel (A), HSPA1A significantly increased IL-1β levels compared to HIRPMI (p<0.0001). However, the combination of HSPA1A and TLR2 BP significantly decreased IL-1β levels compared to HSPA1A alone (p<0.01). Panel (B) shows a significant increase in IL-4 production with HSPA1A treatment alone (p<0.0001), which was markedly reduced with the combination treatment (p<0.0001). Panel (C) and (D) display similar trends for IL-10 and TNF-α, respectively, with significant reductions in the combination treatment group compared to HSPA1A alone (p<0.0001 for IL-10 and p<0.001 for TNF- α).
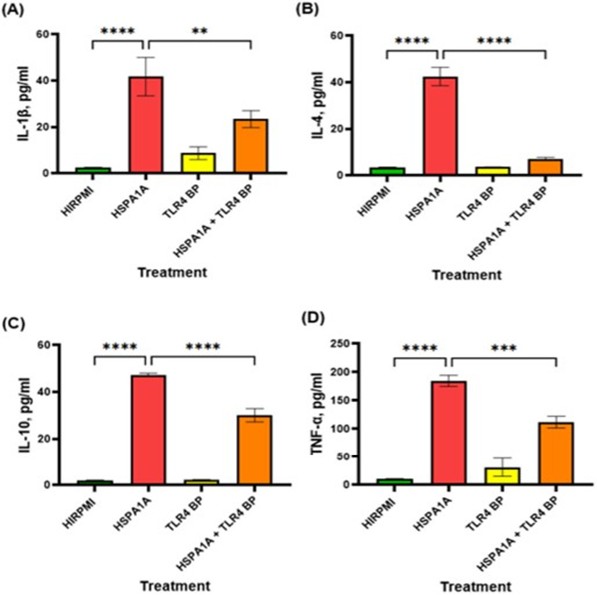
Figure 3: Effects of various treatments on IL-1β, IL-4, IL-10, and TNF-α cytokine production in THP-1 macrophages. The treatments include HIRPMI (control), HSPA1A, TLR4 blocking peptide (TLR4 BP), and a combination of HSPA1A with TLR4 BP. Data are shown as mean concentration (pg/ml) ± SD, n = 3 and tested by Ordinary one-way ANOVA with Bonferroni’s multiple comparison post hoc. Significance levels are indicated as follows: ** (p<0.01), *** (p<0.001), and **** (p<0.0001), comparing treated groups to the control and between specific treatments.
The data in Figure 4 demonstrates the effects of HSPA1A, TLR5 BP, and their combination on cytokine production. In panel (A), treatment with HSPA1A significantly elevated IL-1β levels compared to HIRPMI (p<0.0001). The combination treatment with HSPA1A and TLR5 BP resulted in a significant reduction of IL-1β levels compared to HSPA1A alone (p<0.05). Panel (B) shows that HSPA1A treatment led to a marked increase in IL-4 production compared to control (p<0.0001), while the addition of TLR5 BP significantly reduced IL-4 levels compared to HSPA1A alone (p<0.0001). In panel (C), IL-10 production was significantly upregulated by HSPA1A treatment (p<0.0001), with a significant decrease observed in the combination treatment (p<0.001). Panel (D) reveals that TNF-α levels were significantly higher with HSPA1A treatment (p<0.0001) and were notably reduced with combination treatment (p<0.0001).
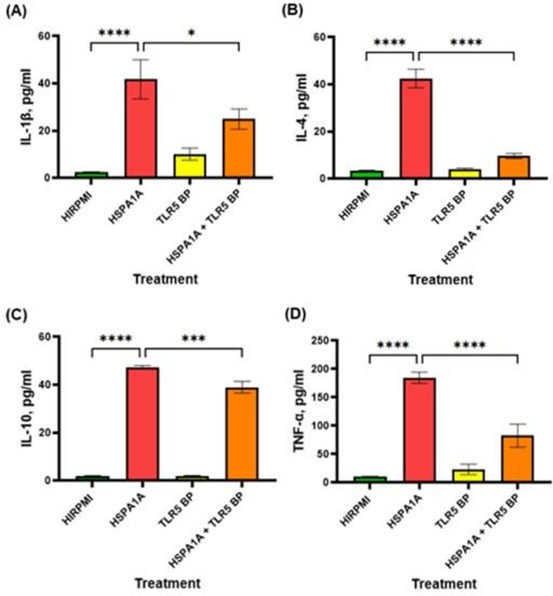
Figure 4: Effects of various treatments on IL-1β, IL-4, IL-10, and TNF-α cytokine production in THP-1 macrophages. The treatments include HIRPMI (control), HSPA1A, TLR5 blocking peptide (TLR5 BP), and a combination of HSPA1A with TLR5 BP. Data are shown as mean concentration (pg/ml) ± SD, n = 3 and tested by Ordinary one-way ANOVA with Bonferroni’s multiple comparison post hoc. Significance levels are indicated as follows: * (p<0.05), *** (p<0.001), and **** (p<0.0001), comparing treated groups to the control and between specific treatments.
The data in Figure 5 demonstrate the effects of HSPA1A, TLR7 blocking peptide (TLR7 BP), and their combination on cytokine production. Panel (A) shows a significant increase in IL-1β levels following HSPA1A treatment compared to HIRPMI (p<0.0001), which is reduced in the combination treatment compared to HSPA1A alone (p<0.05). IL-4 follows a similar pattern as IL-1β (p<0.0001 for both). Panel (C) indicates that HSPA1A significantly elevated IL-10 (p<0.0001), with a reduction in the combination treatment (p<0.0001). Panel (D) shows a similar pattern.
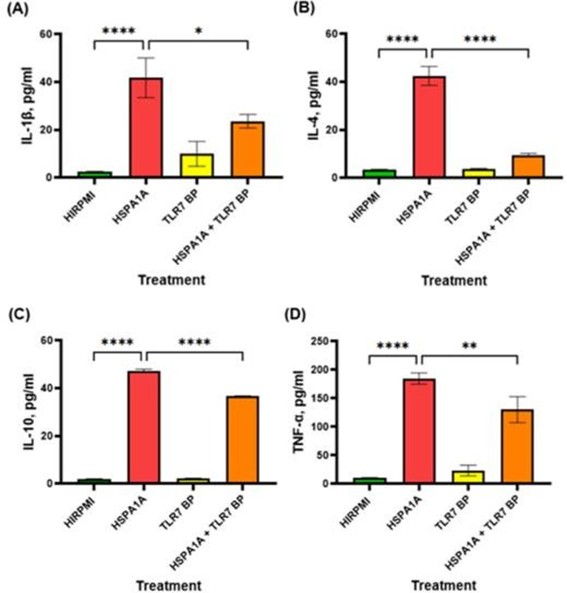
Figure 5: Effects of various treatments on IL-1β, IL-4, IL-10, and TNF-α cytokine production in THP-1 macrophages. The treatments include HIRPMI (control), HSPA1A, TLR7 blocking peptide (TLR7 BP), and a combination of HSPA1A with TLR7 BP. Data are shown as mean concentration (pg/ml) ± SD, n = 3 and tested by Ordinary one-way ANOVA with Bonferroni’s multiple comparison post hoc. Significance levels are indicated as follows: * (p<0.05), *** (p<0.01), and **** (p<0.0001), comparing treated groups to the control and between specific treatments.
The analysis in Figure 6 illustrates the impact of HSPA1A, various TLR BP and their combination on MyD88 signaling molecule in cytokine production. In panel A, the concentration of MyD88 significantly decreased following HSPA1A treatment compared to the HIRPMI (p<0.001). The combination of HSPA1A with TLR2 BP resulted in a significantly higher MyD88 concentration compared to HSPA1A alone (p<0.0001). In B, MyD88 levels significantly decreased with HSPA1A treatment compared with HIRPMI (p<0.0001). The combination treatment further significantly reduced MyD88 levels (p<0.01). Panel C shows that MyD88 levels significantly decreased in the HSPA1A-treated group compared to HIRPMI (p<0.05). However, no significant difference was observed between the HSPA1A and HSPA1A + TLR5 BP treatments. In D, HSPA1A treatment resulted in a significant decrease in MyD88 levels compared to the HIRPMI control (p<0.001). The combination treatment shows further decrease in MyD88 levels compared to HSPA1A alone (p<0.0001).
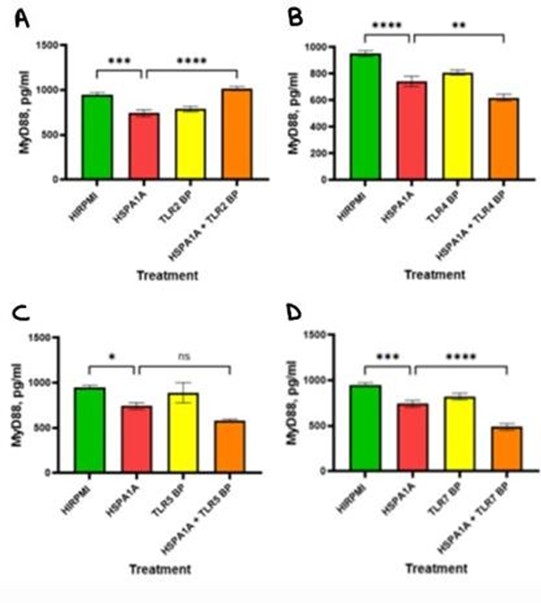
Figure 6: Concentration levels of MyD88 in THP-1 macrophages treated with HIRPMI (control), TLR2 blocking peptide (TLR2 BP) (panel A), TLR4 blocking peptide (TLR4 BP) (panel B), TLR5 blocking peptide (TLR5 BP) (panel C), TLR7 blocking peptide (TLR7 BP) (panel D), and a combination of HSPA1A and blocking peptides. The data are represented as mean concentrations (pg/ml or absorbance at 450 nm) ± SD, n = 3 and tested by Ordinary one-way ANOVA with Bonferroni’s multiple comparison post hoc. Significance levels are indicated as follows: ns (p>0.05), * (p<0.05), ** (p<0.01), *** (p<0.001), and **** (p<0.0001).
The data in Figure 7 shows the effects of HSPA1A, various TLR BP and their combination on MAPK p38 signaling molecule in cytokine production. Panel A shows that MAPK p38 levels significantly increased in the HSPA1A-treated group compared to the HIRPMI control (p<0.0001). However, treatment with combination of HSPA1A with TLR2 BP significantly reduced the MAPK p38 levels (p<0.0001). Panel B shows that MAPK p38 levels increased significantly with HSPA1A treatment compared to control (p<0.0001), and this increase was even bigger with combination treatment (p<0.0001). In panel C, MAPK p38 levels significantly increased in the HSPA1A + TLR5 BP group compared to HSPA1A alone (p<0.01), and a significant increase in HSPA1A treatment compared to HIRPMI (p<0.01). Panel D shows that HSPA1A alone increased MAPK p38 levels significantly (p<0.0001), while the combination treatment further elevated these levels (p<0.001).
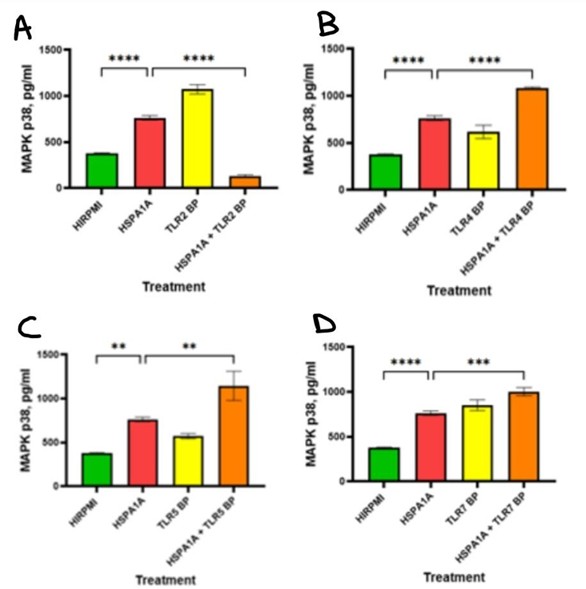
Figure 7: Concentration levels of MAPK in THP-1 macrophages treated with HIRPMI (control), TLR2 blocking peptide (TLR2 BP) (panel A), TLR4 blocking peptide (TLR4 BP) (panel B), TLR5 blocking peptide (TLR5 BP) (panel C), TLR7 blocking peptide (TLR7 BP) (panel D), and a combination of HSPA1A and blocking peptides. The data are represented as mean concentrations (pg/ml or absorbance at 450 nm) ± SD, n = 3 and tested by Ordinary one-way ANOVA with Bonferroni’s multiple comparison post hoc. Significance levels are indicated as follows: ** (p<0.01), *** (p<0.001), and **** (p<0.0001).
The below Figure 8 illustrates the effects of HSPA1A, various TLR BPs, and their combination on NF-κB p65 signaling molecule in cytokine production. In panel A, NF-κB p65 levels were not significantly different between HIRPMI and HSPA1A treatment (p>0.05), however the combination of HSPA1A with TLR2 increased NF-κB p65 levels compared to HSPA1A alone (p<0.01) or TLR2 alone. In panel B, NF-κB p65 levels showed similar pattern as Figure 6. Panel C indicates a slight but significant increase in NF-κB p65 levels in the combination treatment group compared to HSPA1A (p<0.05) alone or TLR5 alone, with no significant differences observed between the HSPA1A and HIRPMI treatments. In panel D, no significant difference was observed in NF-κB p65 levels between HIRPMI and HSPA1A treatments (p>0.05). However, the combination of HSPA1A with TLR7 BP significantly increased NF-κB p65 levels compared to HSPA1A alone (p<0.01).
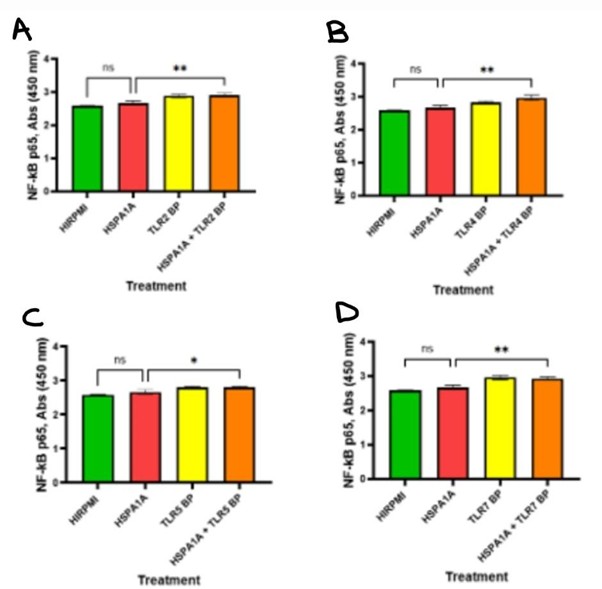
Figure 8: Concentration levels of NF-κB p65 in THP-1 macrophages treated with HIRPMI (control), TLR2 blocking peptide (TLR2 BP) (panel A), TLR4 blocking peptide (TLR4 BP) (panel B), TLR5 blocking peptide (TLR5 BP) (panel C), TLR7 blocking peptide (TLR7 BP) (panel D), and a combination of HSPA1A and blocking peptides. The data are represented as mean concentrations (pg/ml or absorbance at 450 nm) ± SD, n = 3 and tested by Ordinary one-way ANOVA with Bonferroni’s multiple comparison post hoc. Significance levels are indicated as follows: ns (p>0.05), * (p<0.05), and ** (p<0.01).
The data in Figure 9 illustrates the fold change difference in NF-κB expression levels in THP1 macrophage following various treatments. Across all panels (A-D), it is observed that treatment with HSPA1A increased NF-κB expression compared to the control (HIRPMI), but these changes are not statistically significant (p>0.05). The presence of TLR blocking peptides (TLR2 BP, TLR4 BP, TLR5 BP, TLR7 BP) alone or in combination with HSPA1A does not show a notable impact on NF-κB expression levels (p>0.05).
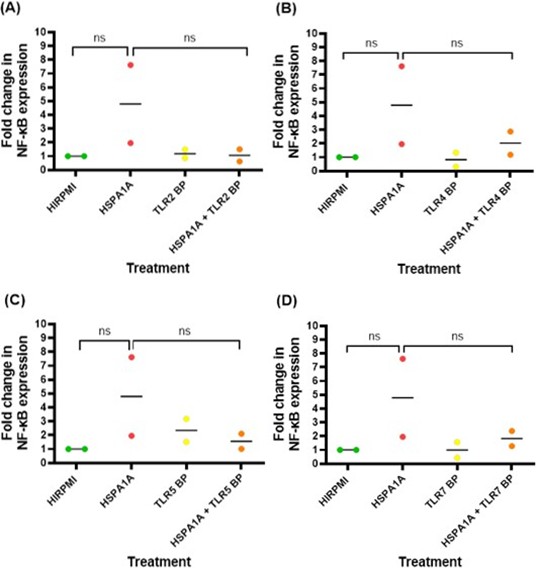
Figure 9: The fold change in NF-κB expression in response to different treatments. The treatments include HIRPMI (control), HSPA1A, TLR2 blocking peptide (TLR2 BP), and a combination of HSPA1A and TLR2 BP. Similarly, with TLR4 BP, TLR5 BP, and TLR7 BP. The fold change is calculated relative to the control (HIRPMI), which is set as 1. Data points represent individual biological replicates, and the horizontal bar represent the mean fold change. Data was analyzed by nonparametric Mann-Whitney test and statistical significance is reported as follows: ns (p>0.05).
Discussion
This study investigated the role of endogenous extracellular HSPs in modulating cytokine secretion by utilizing exogenous recombinant HSPA1A as a model. This approach allowed us to examine cytokine secretion levels (IL-1β, IL-4, IL-10, and TNF- α) in PMA-differentiated THP-1 macrophages under controlled experimental conditions, mimicking the effects of endogenous extracellular HSPA1A. Treatment with 1000 ng/ml of recombinant HSPA1A significantly increased the secretion of all four cytokines (p<0.0001), indicating that HSPA1A plays a critical role in activating the immune system. Notably, there was a consistent and significant increase in the production of both pro-inflammatory cytokines (IL-1β and TNF-α) and anti- inflammatory cytokines (IL-4 and IL-10) (p<0.0001), which aligns with previous research [18], suggesting HSPA1A stimulates cytokine production via TLR interactions.
In this study, the use of TLR-specific blocking peptide provided critical insights into the involvement of these receptors in HSPA1A-induced cytokine production. Blocking TLR2, TLR4, TLR5 and TLR7 significantly decreased the cytokine responses induced by HSPA1A, emphasizing the key role of these receptors. These findings highlight the dual role of HSPA1A in promoting both pro-inflammatory and anti-inflammatory responses, providing new insights into the complex mechanisms in which HSPA1A modulates the immune system.
This dual role suggests that HSPA1A functions both as a danger signal and a modulator of immune system [4]. The ability of HSPA1A to elicit both pro- and anti-inflammatory cytokines appears to be influenced by both receptor-specific activation and the immune context [18,40]. For instance, activation of TLR2 by HSPA1A is generally associated with anti-inflammatory responses through exclusive engagement of MyD88-dependent signaling, whereas TLR4 activation predominantly drives pro-inflammatory pathways via both MyD88 and TRIF-dependent pathways [18]. Furthermore, TLR5 and TLR7 may modulate cytokine responses directly, possibly through cross-talk with MAPK or interferon pathways [41,42]. These findings suggest that specific TLR pathways are differentially activated by HSPA1A, explaining the observed difference in cytokine production.
Recombinant HSPA1A treatment of PMA-differentiated THP-1 macrophages significantly increased both pro-inflammatory and anti-inflammatory cytokines (Figure 1). There was a significant increase in IL-1β and TNF-α levels, both pro-inflammatory cytokines, compared to control (p<0.0001). These findings align with previous research indicating that HSPs, such as HSPA1A, can activate immune system, enhancing inflammatory responses [11]. The ability of HSPA1A to induce proinflammatory cytokines supports its potential role in the pathogenesis of chronic inflammatory diseases, as these cytokines are often implicated in the exacerbation of inflammatory responses [7]. Interestingly, the results in this study also show a significant increase in anti-inflammatory cytokines IL-4 and IL-10 levels (p<0.0001). These findings imply that HSPA1A might also play a role in promoting resolution of inflammation or in regulating the immune response to prevent excessive damage [33]. These findings align with [34], who reported that extracellular HSPs could induce both pro- and anti-inflammatory responses, although the mechanisms remain unclear.
This study utilized TLR-specific blocking peptides to investigate the role of TLRs in mediating the immune response induced by recombinant HSPA1A in THP-1 macrophages. TLR-blocking peptides have been used in several studies to investigate TLR-mediated cytokine secretion; for instance, Ogbodo et al. [18] observed a significant reduction in cytokine production in U937 monocytic cells using TLR2, TLR4, TLR5, and TLR7 specific blocking peptides. This study focused on TLR2, TLR4, TLR5, and TLR7, receptors that have been observed to be involved in HSP-mediated immune modulation [11]. Other TLRs, such as TLR1, may also contribute to these processes; however, they are not considered primary receptor for HSPs [45]. The use of TLR- blocking peptides has demonstrated high specificity and selectivity in TLR-mediated pathways; for example, blocking peptides specifically targeting TLR4 have been effective in selectively modulating TLR4 signaling pathways and reducing uncontrolled inflammation [36]. Similarly, in this study a significant reduction was observed in IL-1β, IL-4, IL-10, and TNF-α levels upon pre-treatment with TLR2, TLR4, TLR5, and TLR7 blocking peptides, emphasizes the involvement of these receptors in the HSPA1A-induced immune response. Notably, TLR2 and TLR4 blocking peptides had a more substantial impact on cytokine reduction, indicating their critical role in this signaling pathway. In contrast, TLR5 and TLR7 have a minor role in cytokine production. These findings align with previous observations that TLR2 and TLR4 are more efficient in being recognized by a variety of ligands, including HSPs [13]. In contrast, TLR5 has been observed to mainly recognize bacterial flagellin, and TLR7 to detect single-stranded RNA and they might not be involved in the HSPA1A- mediated immune response and cytokine production [21]. Additionally, the observed reduction in cytokine levels upon blocking individual TLRs in this study suggests that HSPA1A engages these receptors to mediate its effects. However, the partial inhibition of cytokine production observed indicates that synergistic signaling might also contribute [39]. This observation aligns with findings from Asea et al. [11], which demonstrated that extracellular HSPA1A interacts with multiple TLRs, potentially leading to overlapping signaling cascades. The residual cytokine production observed despite the use of TLR-specific blocking peptides could be attributed to compensatory mechanisms within the immune system. Previous studies [22,23] demonstrated that peptide inhibitors can effectively reduce cytokine production by blocking MyD88- dependent TLR signaling pathways. However, this study observed some levels of cytokine production in THP-1 cells treated only with blocking peptides, suggesting that additional pathways or receptors may contribute to the immune response.
This study further investigated the signaling pathways activated by HSPA1A in inducing cytokine production in THP-1 macrophages, focusing on key molecules involved in TLR signaling, such as MyD88, MAPK, and NF-κB. HSPA1A binding to TLRs initiate a signaling cascade that activates NF-κB and MAPK pathways, crucial for regulating cytokine production and immune response [7]. ELISA was utilized to quantify the level of MyD88, NF-κB, and MAPK THP-1 macrophages following HSPA1A treatment and TLR-specific blocking peptides.
Figures 6-8 indicate that HSPA1A treatment significantly alters these signaling molecules. Interestingly, MyD88 levels were higher in control samples (HIRPMI) than in those treated with HSPA1A, possibly due to feedback mechanisms regulating MyD88 levels post-activation [19]. As MyD88 is one of the first molecules activated in these pathways, its reduced levels after 18 hours suggest downregulation due to degradation or modification, consistent with negative feedback mechanisms [20]. This observation aligns with findings that these signaling pathways are tightly controlled to prevent excessive inflammatory responses, which could lead to pathological conditions [24].
MyD88 initiates several signaling cascades, including those activating MAPK and NF-κB, essential for proinflammatory cytokine transcription [25]. This study showed that MAPK activation, specifically the p38 isoform, was higher in HSPA1A-treated cells and in combination treatments compared to control, suggesting a significant role for the MAPK pathway in HSPA1A-induced cytokine production. The increased activation of MAPK p38 supports its involvement in mediating inflammatory responses through the synthesis and release of pro-inflammatory cytokines, such as IL-1β and TNF-α [26].
Surprisingly, NF-κB p65 activity was elevated in all treatment groups, including controls, indicating that THP-1 macrophages might maintain a baseline level of NF-κB activation, possibly due to their differentiation state or inherent cellular stress [27]. Although the elevation of NF-κB p65 in response to HSPA1A was not statistically significant (p>0.05), it suggested a trend towards increased activation, indicating that HSPA1A can enhance NF-κB activity, potentially raising cytokine expression levels [28]. Figure 8 demonstrates that combination treatments with HSPA1A and specific TLR-blocking peptides result in higher NF-κB activation compared to either HSPA1A or TLR stimulation alone. This increase likely reflects a compensatory response, where partial inhibition of specific TLR pathways redirects signaling through other unblocked TLR’s, amplifying NF-κB levels [46].
It is important to note that components of the NF-κB signaling pathway, such as the p65 subunit detected in this study, go through post-translational modifications (PTMs), including phosphorylation and ubiquitination [29]. These modifications can affect the stability, localization, and activity of the NF-κB complex, complicating the detection of active p65 after 18 hours of HSPA1A treatment [29]. While the detection of the p65 subunit is common due to its significant role in NF-κB signaling, other subunits like p50 could also be differentially regulated and contribute to the cytokine production observed in this study [30].
Asea et al. [11] demonstrated that HSPA1A can induce a potent cytokine activity by binding to plasma membrane and activating NF-κB via the MyD88/ NF-κB signaling pathway. However, TLR4 can also use the alternative adaptor protein TRIF to activate TRAF6, leading to NF-κB induction and cytokine release [31]. Therefore, reduced MyD88 activation does not necessarily preclude NF-κB activation, explaining why this study did not detect elevated levels of MyD88 but did detect NF-κB and elevated cytokines.
The analysis of NF-κB expression revealed further insights into the transcriptional activity of this signaling pathway. Although the overall increase in NF-κB expression following HSPA1A treatment was not statistically significant (p>0.05), variations in fold change among biological replicates were observed. One replicate exhibited a notably high fold change (Figure 9), suggesting that there may be significant variability in the cellular response to HSPA1A due to differences in the activation state of the cells or variations in the expression of co-receptors or adaptor proteins that modulate NF-κB activity [32].
The role of HSPA1A in modulating cytokine production offers promising therapeutic potential. However, it also poses risks of unintended pro-inflammatory effects, particularly in conditions where immune system is already compromised, such as autoimmune or chronic inflammatory diseases. Overactivation of HSPA1A-mediated immune pathways could further aggravate these conditions. Therefore, a detailed understanding of the underlying mechanism is crucial to developing controlled and precise strategies for future therapeutic applications.
This study primarily focused on IL-1β, IL-4, IL-10 and TNF-α cytokines. While these are critical markers, future studies should include additional cytokines and multiple detection time points to provide a more comprehensive understanding of HSPA1A- mediated immune responses. The response observed in this study could be due to the specific concentrations of HSPA1A and TLR-specific blocking peptides used, as these concentrations were selected based on prior studies demonstrating its effectiveness in modulating cytokine secretion and inhibiting TLR-mediated pathways [18,37]. While these concentrations resulted in significant findings and have been reported as non-toxic [37,38], future studies should include cytotoxicity assessments to confirm their safety across different concentrations of HSPA1A and TLR-blocking peptides [44]. Moreover, while this study thoroughly focused on MyD88-dependent pathways, it did not explore any non MyD88-dependent pathways, such as TRIF or P13K/AKT pathways. Future studies should examine these pathways to gain a more comprehensive understanding of the mechanisms underlying HSPA1A-induced immune responses. Furthermore, this study would have benefitted with the inclusion of western blot analysis to validate the expression and activity of the signaling molecules discussed. Incorporating such analysis in future studies would provide a more comprehensive understanding of the signaling pathways involved.
Conclusion
This study observed that recombinant HSPA1A can activate the immune system, resulting in the secretion of cytokines, specifically IL-1β, IL-4, IL-10, and TNF-α. This activation is mediated through TLRs, particularly TLR2, TLR4, TLR5, and TLR7, highlighting their significant role in HSPA1A-induced immune response. The involvement of key signaling pathways, such as MyD88, MAPK, and NF-κB, suggests a complex regulatory mechanism that controls the immune response. These findings provide valuable insights into the role of HSPA1A and TLR interactions in immune modulation, suggesting their potential as diagnostic and therapeutic targets [2].
Authorship Contribution Statement
EO and HM conceived of the study and designed the experiments. EO and HM supervised the study. NJH executed the experiments and analyzed the data. EO, HM, RKE, and CRW supported in the experiments. NJH, EO, and HM interpreted the data and planned the publication. NJH wrote the first draft of the manuscript, and EO edited and approved the manuscript.
Declaration of Competing Interest
None
Acknowledgments
School of Life Sciences, Coventry University, UK
References
2. Hu C, Yang J, Qi Z, Wu H, Wang B, Zou F, et al. Heat shock proteins: Biological functions, pathological roles, and therapeutic opportunities. MedComm (2020). 2022 Aug 2;3(3):e161.
3. Lianos GD, Alexiou GA, Mangano A, Mangano A, Rausei S, Boni L, Dionigi G, Roukos DH. The role of heat shock proteins in cancer. Cancer Lett. 2015 May 1;360(2):114-8.
4. Wheeler DS, Wong HR. Heat shock response and acute lung injury. Free Radic Biol Med. 2007 Jan 1;42(1):1-14.
5. Koliński T, Marek-Trzonkowska N, Trzonkowski P, Siebert J. Heat shock proteins (HSPs) in the homeostasis of regulatory T cells (Tregs). Cent Eur J Immunol. 2016;41(3):317-23.
6. Robert J. Evolution of heat shock protein and immunity. Dev Comp Immunol. 2003 Jun-Jul;27(6-7):449-64.
7. Tsan MF, Gao B. Heat shock proteins and immune system. J Leukoc Biol. 2009 Jun;85(6):905-10.
8. Moroi Y, Mayhew M, Trcka J, Hoe MH, Takechi Y, Hartl FU, et al. Induction of cellular immunity by immunization with novel hybrid peptides complexed to heat shock protein 70. Proc Natl Acad Sci U S A. 2000 Mar 28;97(7):3485-90.
9. McCallister C, Kdeiss B, Nikolaidis N. HspA1A, a 70-kDa heat shock protein, differentially interacts with anionic lipids. Biochem Biophys Res Commun. 2015 Nov 27;467(4):835-40.
10. Klink M, Nowak M, Kielbik M, Bednarska K, Blus E, Szpakowski M, et al. The interaction of HspA1A with TLR2 and TLR4 in the response of neutrophils induced by ovarian cancer cells in vitro. Cell Stress Chaperones. 2012 Nov;17(6):661-74.
11. Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002 Apr 26;277(17):15028-34.
12. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014 Sep 25;5:461.
13. Sameer AS, Nissar S. Toll-Like Receptors (TLRs): Structure, Functions, Signaling, and Role of Their Polymorphisms in Colorectal Cancer Susceptibility. Biomed Res Int. 2021 Sep 12;2021:1157023.
14. Asea A. Heat shock proteins and toll-like receptors. Handb Exp Pharmacol. 2008;183(183):111-27.
15. Saito K, Dai Y, Ohtsuka K. Enhanced expression of heat shock proteins in gradually dying cells and their release from necrotically dead cells. Exp Cell Res. 2005 Oct 15;310(1):229-36.
16. Wu FH, Yuan Y, Li D, Liao SJ, Yan B, Wei JJ, et al. Extracellular HSPA1A promotes the growth of hepatocarcinoma by augmenting tumor cell proliferation and apoptosis-resistance. Cancer Lett. 2012 Apr 28;317(2):157-64.
17. Chiricosta L, Gugliandolo A, Bramanti P, Mazzon E. Could the Heat Shock Proteins 70 Family Members Exacerbate the Immune Response in Multiple Sclerosis? An in Silico Study. Genes (Basel). 2020 Jun 3;11(6):615.
18. Ogbodo E, Michelangeli F, Williams JHH. Exogenous heat shock proteins HSPA1A and HSPB1 regulate TNF-α, IL-1β and IL-10 secretion from monocytic cells. FEBS Open Bio. 2023 Oct;13(10):1922-40
19. Saikh KU. MyD88 and beyond: a perspective on MyD88-targeted therapeutic approach for modulation of host immunity. Immunol Res. 2021 Apr;69(2):117-28.
20. Liu M, Kang W, Hu Z, Wang C, Zhang Y. Targeting MyD88: Therapeutic mechanisms and potential applications of the specific inhibitor ST2825. Inflamm Res. 2023 Nov;72(10-11):2023-36.
21. Wu H, Wang H, Xiong W, Chen S, Tang H, Han D. Expression patterns and functions of toll-like receptors in mouse sertoli cells. Endocrinology. 2008 Sep;149(9):4402-12.
22. Aftabizadeh M, Li YJ, Zhao Q, Zhang C, Ambaye N, Song J, et al. Potent antitumor effects of cell-penetrating peptides targeting STAT3 axis. JCI Insight. 2021 Jan 25;6(2):e136176.
23. Toshchakov VY, Fenton MJ, Vogel SN. Cutting Edge: Differential inhibition of TLR signaling pathways by cell-permeable peptides representing BB loops of TLRs. J Immunol. 2007 Mar 1;178(5):2655-60.
24. Fu Q, Jiang J, Li X, Zhai Z, Wang X, Li C, et al. Activation of MyD88-Dependent TLR Signaling Modulates Immune Response of the Mouse Heart during Pasteurella multocida Infection. Microorganisms. 2023 Feb 4;11(2):400.
25. Bayer AL, Alcaide P. MyD88: At the heart of inflammatory signaling and cardiovascular disease. J Mol Cell Cardiol. 2021 Dec;161:75-85.
26. Ganguly P, Macleod T, Wong C, Harland M, McGonagle D. Revisiting p38 Mitogen-Activated Protein Kinases (MAPK) in Inflammatory Arthritis: A Narrative of the Emergence of MAPK-Activated Protein Kinase Inhibitors (MK2i). Pharmaceuticals (Basel). 2023 Sep 12;16(9):1286.
27. Chanput W, Mes JJ, Wichers HJ. THP-1 cell line: an in vitro cell model for immune modulation approach. Int Immunopharmacol. 2014 Nov;23(1):37-45.
28. Wen Q, Zhan B, Jin L, Peng Z, Liu J, Zhu L, Yang D, Xu X, Zhang L, Li G, Zhao Z. Chlojaponilactone B Attenuates THP-1 Macrophage Pyroptosis by Inhibiting the TLR/MyD88/NF-κB Pathway. Pharmaceuticals (Basel). 2024 Mar 21;17(3):402.
29. Yu X, Li W, Deng Q, Liu H, Wang X, Hu H, et al. MYD88 L265P elicits mutation-specific ubiquitination to drive NF-κB activation and lymphomagenesis. Blood. 2021 Mar 25;137(12):1615-27.
30. Baud V, Collares D. Post-Translational Modifications of RelB NF-κB Subunit and Associated Functions. Cells. 2016 May 4;5(2):22.
31. Deguine J, Barton GM. MyD88: a central player in innate immune signaling. F1000Prime Rep. 2014 Nov 4;6:97
32. Dorrington MG, Fraser IDC. NF-κB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front Immunol. 2019 Apr 9;10:705
33. Borges TJ, Wieten L, van Herwijnen MJ, Broere F, van der Zee R, Bonorino C, van Eden W. The anti-inflammatory mechanisms of Hsp70. Front Immunol. 2012 May 4;3:95.
34. Bhatia S, Diedrich D, Frieg B, Ahlert H, Stein S, Bopp B, et al. Targeting HSP90 dimerization via the C terminus is effective in imatinib-resistant CML and lacks the heat shock response. Blood. 2018 Jul 19;132(3):307-20.
35. Chandrasekar V, Panicker AJ, Dey AK, Mohammad S, Chakraborty A, Samal SK, et al. Integrated approaches for immunotoxicity risk assessment: challenges and future directions. Discover Toxicology. 2024 Oct 29;1(1):9.
36. Buccini DF, Roriz BC, Rodrigues JM, Franco OL. Antimicrobial peptides could antagonize uncontrolled inflammation via Toll-like 4 receptor. Front Bioeng Biotechnol. 2022 Dec 7;10:1037147.
37. Piao W, Shirey KA, Ru LW, Lai W, Szmacinski H, Snyder GA, et al. A Decoy Peptide that Disrupts TIRAP Recruitment to TLRs Is Protective in a Murine Model of Influenza. Cell Rep. 2015 Jun 30;11(12):1941-52.
38. Figueiredo C, Wittmann M, Wang D, Dressel R, Seltsam A, Blasczyk R, et al. Heat shock protein 70 (HSP70) induces cytotoxicity of T-helper cells. Blood, The Journal of the American Society of Hematology. 2009 Mar 26;113(13):3008-16.
39. Huang Y, Zhang Q, Lubas M, Yuan Y, Yalcin F, Efe IE, et al. Synergistic toll-like receptor 3/9 signaling affects properties and impairs glioma-promoting activity of microglia. Journal of Neuroscience. 2020 Aug 12;40(33):6428-43.
40. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010 Mar 19;140(6):805-20.
41. Petes C, Odoardi N, Gee K. The Toll for Trafficking: Toll-Like Receptor 7 Delivery to the Endosome. Front Immunol. 2017 Sep 4;8:1075.
42. Peroval MY, Boyd AC, Young JR, Smith AL. A critical role for MAPK signalling pathways in the transcriptional regulation of toll like receptors. PLoS One. 2013;8(2):e51243.
43. Singh AV, Bhardwaj P, Laux P, Pradeep P, Busse M, Luch A, et al. AI and ML-based risk assessment of chemicals: predicting carcinogenic risk from chemical-induced genomic instability. Front Toxicol. 2024 Nov 26;6:1461587.
44. Singh AV, Kayal A, Malik A, Maharjan RS, Dietrich P, Thissen A, et al. Interfacial Water in the SARS Spike Protein: Investigating the Interaction with Human ACE2 Receptor and In Vitro Uptake in A549 Cells. Langmuir. 2022 Jul 5;38(26):7976-88.
45. Kaur P, Asea A. Toll-Like Receptors and Infectious Diseases: Role of Heat Shock Proteins. In: Pockley A, Calderwood S, Santoro M, (eds). Prokaryotic and Eukaryotic Heat Shock Proteins in Infectious Disease. Heat Shock Proteins, vol 4. Dordrecht: Springer. 2009.
46. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. 2015;33:257-90.