Abstract
The recently published review by Baeten et al. outlines the scientific and translational evidence supporting the role of cyclin dependent kinase (CDK) 4/6 in rheumatoid arthritis (RA) pathology, and it discusses how this stromal (synovial fibroblast) target provides the basis for a drug class with a novel non-immunosuppressive mechanism of action that could change the current immunosuppressive treatment paradigm in RA. The early clinical trial (phase 1b) of the next generation CDK4/6 inhibitor, ELV001, showed the rapid clinical improvement, and ELV001 was well tolerated with no clinically meaningful safety signals. This warrants further clinical evaluation of ELV001 in a larger group of patients with active RA, and the phase 2 study with ELV001 (START SYNERGY) is currently in progress.
This commentary article on the published review by Baeten et al. describes additional perspectives, in particular, what is the next step in the clinical development of CDK4/6 inhibition for RA, as well as where CDK4/6 inhibition could be positioned in the RA treatment algorithm. A novel combination strategy that pairs systemic immunosuppression (current therapeutic options) with a synovial-targeted non-immunosuppressive anti-rheumatic therapy (with ELV001) is expected to leverage orthogonal mechanisms of action, and this approach would represent a distinct therapeutic paradigm for RA.
Keywords
Rheumatoid arthritis, Fibroblast-like synoviocyte, Cyclin dependent kinase, Non-immunosuppressive mechanism of action, ELV001
Commentary
The recently published article entitled “Targeting fibroblast-like synoviocytes with CDK4/6 inhibitors as a novel, non-immunosuppressive treatment strategy for rheumatoid arthritis” [1] outlines the scientific, and translational evidence supporting the role of cyclin dependent kinase (CDK) 4/6 in rheumatoid arthritis (RA) pathobiology. It discusses how this stromal target provides the basis for a drug class with a novel mechanism of action which could change the current immunosuppressive treatment paradigm in RA. Finally, it flags the safety concerns related to existing CDK4/6 inhibitors that are approved for oncology indications and discusses how the next generation CDK4/6 inhibitors with improved therapeutic index – as exemplified by TCK-276 (now also known as ELV001) – are now moving into clinical development in RA [2–4].
Why We Need Novel Therapies in RA
RA is a prevalent chronic immune-mediated inflammatory disease caused by an autoimmune attack of the synovial joints of the appendicular skeleton. The disease pathology is characterized by activation of autoreactive T and B cells leading to chronic inflammation in the stromal compartment of synovial joints. Accordingly, the current treatment approaches primarily focus on altering adaptive or innate immune responses by targeting T cells, B cells, or pro-inflammatory cytokines. Despite the availability of a variety of targeted therapies across multiple drug classes, including tumor necrosis factor (TNF) inhibition, interleukin-6 (IL-6) inhibition, co-stimulation blockade, B cell depletion, and Janus kinase (JAK) inhibition, most patients still fail to reach remission. A commonly encountered example is patients who receive TNF inhibitors where 40–50% of patients demonstrate partial response but fail to reach low disease activity or remission. In addition, there are a group of patients who are entirely refractory to immunosuppressive therapies [5–8]. Regarding a new immunosuppressive therapy candidate, for instance IL-17 inhibition, limited clinical efficacy by IL-17 blockade in RA was reported; possible reasons for the limited clinical success is summarized in a review by Taams [9].
A series of recent studies have defined this "difficult-to-treat RA" (D2TRA) patient phenotype and outlined the significant unmet medical need [10]. Interestingly, single cell RNA sequencing studies performed on synovial biopsies of patients with an insufficient response to TNF inhibition, IL-6 and B cell depletion consistently showed a pronounced fibroblast signature [11–13] indicating that fibroblast-like synoviocytes (FLSs) in RA are not merely passive responders to inflammation but play a central role in the pathobiology of RA synovitis. Very recently, similar findings were reported in newly diagnosed RA patients who failed to achieve remission when initiated on either triple classical disease-modifying anti-rheumatic drug (DMARD) therapy or TNF inhibition [14]. Moreover, a fibroblast activation protein inhibitor (FAPI)-PET CT study in healthy individuals at risk of developing RA showed that FLS activation occurs before the onset of RA and influences the risk of developing disease [15]. Collectively, these new data support the concept that FLS do not only contribute to refractory disease but are actively involved in all stages of RA.
The increasing recognition of the role of FLSs in RA (RA-FLSs) in the disease process has led to the hypothesis that current therapeutic options targeting immune cells may be insufficient to achieve deep and long-lasting remission in RA and therefore that novel drugs targeting specifically target RA-FLSs could be a valuable addition to our therapeutic armamentarium [16,17].
Why We Should Target RA-FLSs
First, RA-FLSs are implicated in many pathological mechanisms relating to joint destruction. Chronically activated and epigenetically transformed RA-FLSs produce cartilage matrix-degrading enzymes such as metalloproteinases (MMPs) and express receptor activator of nuclear factor κB (RANK) ligand, which drive RANK-expressing osteoclasts to erode the peri-articular bone [18,19]. Second, RA-FLSs contribute directly to chronic synovial inflammation by producing a variety of pro-inflammatory factors which act in an autocrine and paracrine manner on resident stromal cells and infiltrating immune cells in the synovium [20–22]. A third and emerging pathobiological feature of RA-FLSs is their ability to attract, activate and support autoimmune T and B cells [23–34].
Although RA-FLSs have been proposed as a potential non-immune therapeutic target [35–37], this approach is still in its infancy. Cadherin-11 and integrin-α9 have been pursued as potential FLS targets but both experimental drugs failed in phase 2 trials [38,39]. This could be potentially explained by a suboptimal pharmacokinetic/pharmacodynamic profile impairing antibody diffusion to the target synovial tissue.
Why Target CDK4/6
The review by Baeten et al. [1] describes why CDK4/6 could be an attractive novel stromal target for the treatment of RA. Although a variety of CDKs are reported to be potentially involved in the pathobiology of RA, CDK4/6 are considered the most promising targets based on genetics [40–42], mode of action (MoA) [43–46], efficacy in in vitro experiments and in vivo arthritis models [43,47–51], as well as translational and clinical data [52–55]. Importantly, CDK4/6 are not only key cell cycling regulators that govern the proliferation of FLSs but are also critical transcriptional regulators that potentiate transcription factor AP1 and nuclear factor κB signaling in RA-FLSs. Accordingly, blocking CDK4/6 inhibited both the proliferation and the aggressive phenotype (e.g. production of MMP3, granulocyte-monocyte colony stimulating factor) of RA-FLSs in vitro and in vivo. Two key findings from nonclinical in vivo experiments with CDK4/6 inhibition in arthritis models are that this approach, a) does not impair the acquired immune system, as evidenced by unaltered T and B cell responses to type II collagen in the collagen-induced arthritis (CIA) model, and b) acts synergistically with immunosuppressive treatments such as TNF inhibition. A combination with anti-IL-6 was also evaluated in the CIA model. CDK4/6 inhibitor monotherapy significantly decreased the arthritis score and combination with anti-IL-6 further improved histological and radiographic scores [51]. This positions CDK4/6 inhibition as an attractive combination (with either TNF inhibition or IL-6 inhibition) treatment for RA.
Why CDK4/6 Inhibitors Approved in Oncology Have Not Been Tested in RA
To date, several CDK4/6 inhibitors such as palbociclib, ribociclib and abemaciclib have been approved for the treatment of HR+/HER2- advanced breast cancer. Despite case reports of beneficial effects on concurrent RA, these drugs have never been prospectively assessed in this disease. The main reason is the safety and tolerability profile of these drugs which – although acceptable for oncology – is not compatible with chronic treatment of non-life-threatening diseases such as RA. The main side effect of the approved CDK4/6 inhibitors is myelosuppression, with up to 80% of treated patients developing reversible but profound (up to 50% grade 3 or 4) neutropenia within 1 to 2 weeks followed by additional blood cell alterations such as anemia, lymphopenia, and thrombopenia. Additional safety concerns include gastrointestinal side effects such as nausea and diarrhea, and the risk of electrocardiogram changes, specifically QT-interval prolongation [56–61]. An improved safety profile is thus a prerequisite for the potential use of CDK4/6 inhibitors in non-oncology-indications such as RA.
Next Generation CDK4/6 Inhibitor Achieves Anti-Arthritic Effects without Causing Myelosuppression
Teijin Pharma (Japan) initiated a focused drug discovery campaign to find a selective and potent CDK4/6 inhibitor with an improved safety profile. The candidate molecule emerging from this campaign – ELV001 – possesses a high selectivity for and potent inhibitory activity against CDK4/6 in vitro. ELV001 inhibited proliferation and MMP3 secretion in RA synovial fibroblasts in vitro and in vivo using the mouse CIA, mouse collagen antibody-induced arthritis and rat adjuvant-induced arthritis models [62]. In all 3 models, ELV001 dose-dependently inhibited clinical arthritis as well as histological joint destruction. Moreover, the combination of ELV001 with anti-TNF-α suppressed arthritic scores more profoundly than monotherapy with either ELV001 or anti-TNF-α. Tsujimoto et al. demonstrated suppressed MMP3 expression in joints in addition to decreased serum MMP3 levels which was associated with amelioration of arthritis scores [63]. ELV001 did not reduce type II collagen IgG levels, suggesting that ELV001 has no impact on the adaptive immune system.
An important feature of ELV001 is its “clean” safety profile. Based on the safety profile of CDK4/6 inhibitors approved in oncology as described in the previous section, off-target effects on non-synovial proliferative compartments would include myelosuppression and gastrointestinal side effect. Nomura et al. reported that myelosuppression was not observed with ELV001 at therapeutically effective dose levels in nonclinical arthritis models [62], and safety margins based on nonclinical animal toxicity study data were estimated at 8-fold and 2-to-5-fold in rodents and monkeys, respectively [2]. As for the gastrointestinal side effect, nonclinical GLP toxicity studies (26-week repeated dose in rats, as well as 39-week repeated dose in monkeys) did not indicate any toxicologically meaningful concerns; no histopathologic findings in the digestive tract in animals dosed with ELV001 at approximately 40-fold or 20-to-40 fold higher exposure-level as compared with the expected clinical efficacy exposure, in rats or monkeys, respectively (Elevara Medicines, London, UK, data on file).
With respects to the other potential off-target effect, i.e., the risk of electrocardiogram changes, there was no inhibitory action observed in in vitro assay using human ether-a-go-go-related gene (hERG) transfected CHO cells [2], and proarrhythmia study in human induced pluripotent stem cell-derived cardiomyocytes revealed a very low risk of ELV001 on cardiac function. In addition, no safety concerns were demonstrated in monkey cardiovascular telemetry studies [62]. The CDK4/6 inhibitors approved in oncology have signals indicating cardiac risks, i.e., hERG inhibition as well as an inhibitory effect on sodium channel. In contrast, ELV001 has no inhibitory effects on hERG or sodium channel, and this is the important molecular background for the improved safety profile in terms of cardiovascular function.
Nonclinical Efficacy and Safety Profile of ELV001 Translate Favorably in Early Clinical Studies
ELV001 has shown a favorable therapeutic index and signs of early clinical efficacy in patients with active RA in a Phase 1b study [2]. Whilst this study was primarily designed to assess safety/tolerability and pharmacokinetics (PK) of ELV001, it revealed a preliminary signal of clinical efficacy as indicated by improvements in disease activity as measured by American College of Rheumatology 20% improvement criteria and European Alliance of Association for Rheumatology disease activity score-28. ELV001 was well tolerated with no clinically meaningful safety signals. Whilst CDK4/6 possess ubiquitous role in cell-cycle regulation and a range of side effects including gastrointestinal side effects as well as myelosuppression were reported regarding the other CDK4/6 inhibitors as described above, potential off-target effects on non-synovial tissues were not reported in the phase 1b study of ELV001. Of specific interest, there were no neutropenia events or alterations in other hematological indices, as well as no QT prolongation. These clinical data are consistent with the favorable therapeutic index of ELV001 in the nonclinical data as compared to the currently approved CDK4/6 inhibitors in an oncology-indication.
Although the underlying reasons for the differentiated safety profile of ELV001 requires further investigation, Tasaki et al. reported a preferential uptake of ELV001 by RA-FLSs over bone marrow cells as well as a differentiated PK profile [2]. ELV001 was preferentially taken up by RA-FLS approximately 5-fold over bone marrow cells, in contrast to palbociclib which is taken up almost equally by both cell types. PK data for ELV001 showed short half-life (overall t1/2 ranged from approximately 6 to 12 hours after dosing) which appears to be significantly shorter than that of the CDK4/6 inhibitors approved in oncology: palbociclib has a half-life of 26 to 27 hours and abemaciclib approximately 18 to 24 hours. The shorter half-life of ELV001 together with its preferential uptakes by RS-FLSs over bone marrow cells may play an important role in avoiding neutropenia and myelosuppression seen with the CDK4/6 inhibitors approved in oncology.
Next Step in the Clinical Development of CDK4/6 Inhibition for RA
Limitations of the phase 1b study of ELV001 include the small study size and the relatively short treatment duration, which should be taken into consideration when interpreting the clinical efficacy signal. Nevertheless, the rapid clinical improvement and the absence of hematopoietic and cardiac side effects in this study warrant further clinical evaluation of ELV001 in a larger group of patients with active RA. The phase 2 study of this next-generation CDK4/6 inhibitor, ELV001, is currently in progress (START SYNERGY, ClinicalTrials.gov ID: NCT07409103) [64].
This phase 2 study will assess the safety and efficacy of ELV001 in RA patients with a partial but incomplete response to the standard of care (SoC) therapies methotrexate (MTX) and TNF inhibitors (anti-TNF). The rationale for choosing 1) this patient population and 2) the combination approach with SoC immunosuppressant drugs is based on insights from nonclinical models where synergy has been observed between this synovial targeting anti-rheumatic treatment (ELV001) and classical immunomodulators (including MTX). Furthermore, since ELV001 is non-immunosuppressive, it can be safely combined with conventional immunosuppressant therapies without concern for immunosuppression and increased in infection risk. Thus, the data support the paradigm of an add-on therapy in patients with a partial but incomplete response to current DMARDs. This study will thus address the safety and tolerability of ELV001 in combination with SoC and assess if adding ELV001 is able to turn a partial response into a deep response; i.e., low disease activity or remission. If the clinical data supports the use of ELV001 as an adjuvant in patients with a partial response to immunosuppressive therapy, this could provide an alternative option to cycling between different available targeted therapies. It is well established that the response rate of biological DMARDs (bDMARDs) reduces according to the number of prior lines of failed bDMARD therapy. Therefore, providing a potential new option that avoids the need to cycle between treatment lines in patients who exhibit a partial response is attractive. If positive clinical evidence emerges for the adjuvant role of ELV001 then this raises the question as to whether ELV001 could be an effective therapy in the treatment of patients with refractory disease/D2TRA.
Previous attempts to combine targeted therapy, as an alternative to sequential drug cycling, have been universally unsuccessful. Combinations such as anti-TNF therapy with IL-1 blockade or cytotoxic T-lymphocyte antigen-4 immunoglobulin-Fc fusion proteins failed to deliver meaningful gains in efficacy and were associated with increased adverse events, particularly serious infections [65]. In contrast, the present study evaluates a novel combination strategy that pairs systemic immunosuppression with a synovial-targeted anti-rheumatic therapy (ELV001), leveraging orthogonal mechanisms of action. This approach represents a distinct therapeutic paradigm, the safety and efficacy of which are being examined in the current phase 2 study.
With regards to a comparative analysis with current bDMARDs, e.g. onset of action, durability of response, and safety including long‐term cardiovascular risk, the phase 2 study may provide a first glimpse on those by an indirect comparison with approved therapies. However, indirect comparison between trials may be susceptible to biases. The real answer to these important questions will come in large, long-term ELV001 phase 3 studies, ideally with a comparator arm included in these trials.
The biomarker framework would be another important aspect remained to be explored in the future. In the phase 1b study of ELV001, whilst a tendency in decreased serum MMP3 levels in subjects with improved disease activity was observed (Elevara Medicines, London, UK, data on file), biomarker measurements were restricted to peripheral blood samples in this phase 1b study. In the next phases of clinical development, biomarker work would focus on FLS. One way to approach this would be to conduct a study with synovial biopsies from patients treated with ELV001. Such biomarker work may help to confirm and understand the MoA and add prospective pharmacodynamic endpoints to validate target engagement in the patient synovial tissue.
Where CDK4/6 Inhibition Could Be Positioned in the RA Treatment Algorithm
Depending on the benefit/risk profile emerging from the current phase 2 study with ELV001, RA-FLS targeted anti-rheumatic therapies may have additional utility beyond the third line (combination with MTX and TNF inhibitor) being evaluated in this clinical study. First, ELV001 may not only be combined with TNF inhibitor but also with other targeted therapies. For instance, it would be worthwhile exploring combinatorial regimens that leverage CDK4/6 mediated RA-FLS inhibition with cytokine‐targeted agents (e.g. IL‐inhibitor) to address residual inflammatory pathways. In particular, a combination of MTX and JAK inhibitor with ELV001 may lead to a fully oral combination therapy. Nonclinical experiments in arthritis models have shown that ELV001 synergizes not only with TNF inhibitor but also with JAK inhibitor, supporting this positioning. Second, if the safety and tolerability profile of ELV001 is confirmed to be favorable, CDK4/6 inhibition may also be positioned in earlier lines of treatment, especially in combination with MTX. This positioning is supported by in vivo nonclinical experiments as well as by the clinical observations from the phase 1b study, in which half of the patients on active treatment were also on background MTX treatment. Whilst its small sample size with a limited dosing period, ELV001 phase 1b study data indicates the strong potential for additional efficacy by adding-on CDK4/6 inhibitor to MTX in MTX incomplete responder with active RA (Figure 1). Finally, based on the previously mentioned synovial biopsy-based single cell RNA sequencing studies, ELV001 may also be tested in refractory/D2TRA patients.
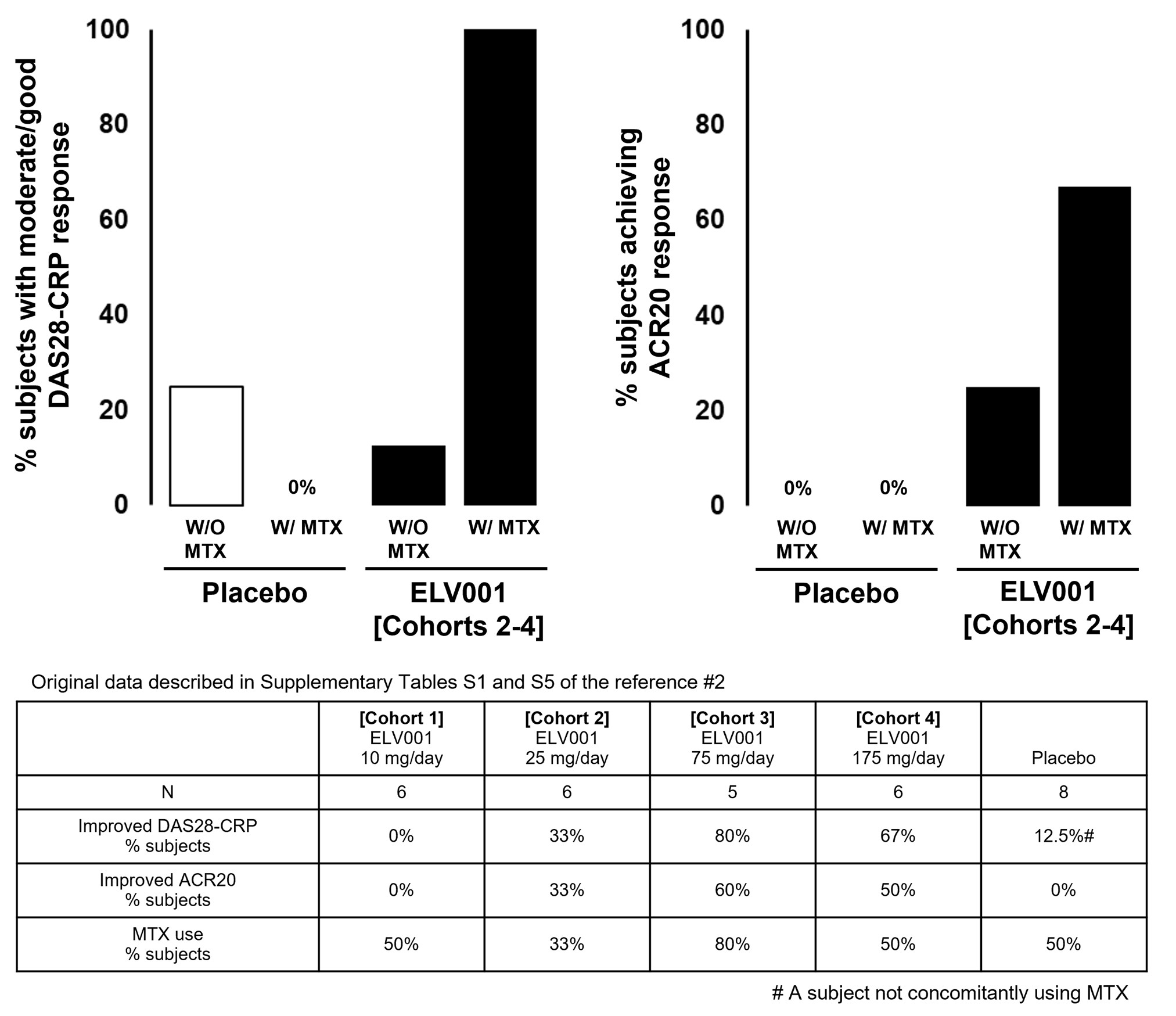
Figure 1. Efficacy of CDK4/6 inhibitor: stratifying by concomitant MTX use. This figure was made by authors using reference #2 (particularly based on the information/data appeared on Supplementally Tables S1 and S5 of the reference). Lower table summarizes the original data described in the reference #2. In the upper graphs, with regards to the subjects treated with ELV001, combined data earned from cohort 2 through 4, which are regarded as an expected efficacy dose-level for ELV001, were additionally analyzed by stratifying with or without MTX concomitant use. DAS28-CRP: Disease Activity Score-28 using CRP; CRP: C-Reactive Protein; ACR20: American College of Rheumatology 20% improvement criteria; MTX: Methotrexate.
Acknowledgements
The authors thank Philip Robinson, MD, PhD, for his critical review and for editorial assistance with this manuscript.
Authors’ Contributions
All authors contributed to writing and editing, and approved the final version of the manuscript for publication.
Disclosure Statements
DB is a consultant to Elevara Medicines. DM was an employee of Teijin Pharma, and is currently a scientific/technical advisor to Elevara Medicines. HK has declared no competing interests for this publication.
Funding
No specific funding was received from any bodies in the public, commercial or not-for-profit sectors to carry out the work described in this article.
References
2. Tasaki D, Tsuruda K, Sun S, Tsumura Y, Asano S, Suzuki Y, et al. A double-blind, placebo-controlled, randomized multiple dose phase 1b trial of a CDK4/6 inhibitor, TCK-276, in patients with active rheumatoid arthritis. Rheumatology (Oxford). 2025; 64 (3): 1036–44.
3. Hosoya T, Saito T, Yasuda S. Cyclin dependent kinase inhibitor: a long-awaited, late-coming, novel class agent in rheumatoid arthritis. Rheumatology (Oxford). 2025; 64 (3): 916–8.
4. Elevara Medicines. Elevara Medicines Raises $70 Million Series A to Advance Phase 2 Rheumatoid Arthritis Trial and Expand Pipeline. England: Elevara Medicines. Avaliable from: https://www.elevara.com/press-releases/22Oct25%20Elevara%20Launch%20and%20Series%20A%20raise.pdf.
5. Tanaka Y. Recent Progress in treatments of rheumatoid arthritis: an overview of developments in biologics and small molecules, and remaining unmet needs. Rheumatology (Oxford). 2021; 60 (Suppl 6): vi12–20
6. Zhao SS, Kearsley-Fleet L, Bosworth A, Watson K, BSRBR-RA Contributors Group, Hyrich KL. Effectiveness of sequential biologic and targeted disease modifying anti-rheumatic drugs for rheumatoid arthritis. Rheumatology (Oxford). 2022; 61 (12): 4678–86.
7. Matsson A, Solomon DH, Crabtree MM, Harrison RW, Litman HJ, Johansson FD. Patterns in the sequential treatment of patients with rheumatoid arthritis starting a biologic or targeted synthetic disease-modifying antirheumatic drug: 10-year experience from a US-based registry. ACR Open Rheumatol. 2024; 6 (1): 5–13.
8. Movahedi M, Kuriya B, Cesta A, Li X, Aydin SZ, Bombardier C, et al. Treatment sequences and lines of therapy in rheumatoid arthritis: a real-world evaluation of retention and effectiveness. Clin Exp Rheumatol. 2025 Nov 12.
9. Taams LS. Interleukin-17 in rheumatoid arthritis: trials and tribulations. J Exp Med. 2020; 217 (3): e20192048.
10. Nagy G, Roodenrijs NMT, Welsing PMJ, Kedves M, Attila Hamar A, van der Goes MC, et al. EULAR definition of difficult- to- treat rheumatoid arthritis. Ann Rheum Dis. 2021; 80 (1): 31–5.
11. Humby F, Durez P, Buch MH, Lewis MJ, Rizvi H, Rivellese F, et al. Rituximab versus tocilizumab in anti-TNF inadequate responder patients with rheumatoid arthritis (R4RA): 16-week outcomes of a stratified, biopsy-driven, multicentre, open-label, phase 4 randomised controlled trial. Lancet. 2021; 397 (10271): 305–17.
12. Rivellese F, Surace AEA, Goldmann K, Sciacca E, Çubuk C, Giorli G, et al. Rituximab versus tocilizumab in rheumatoid arthritis: synovial biopsy-based biomarker analysis of the phase 4 R4RA randomized trial. Nat Med. 2022; 28 (6): 1256–68.
13. Zhang F, Jonsson AH, Nathan A, Millard N, Curtis M, Xiao Q, et al. Deconstruction of rheumatoid arthritis synovium defines inflammatory subtypes. Nature. 2023; 623 (7987): 616–24.
14. Bhamidipati K, McIntyre ABR, Kazerouian S, Ce G, Wong SW, Tran M, et al. Spatial patterning of fibroblast TGFβ signaling underlies treatment resistance in rheumatoid arthritis. Nat Immunol. 2026 Mar;27(3):556–71.
15. Atzinger A, Tascilar K, Kleyer A, Hueber AJ, Schmidkonz C, Kuwert T, et al. Synovial fibroblast activation occurs before the onset of rheumatoid arthritis and influences the risk of developing disease. RMD Open. 2025; 11: e005775.
16. Zou AE, Kongthong S, Muller AA, Brenner MB. Fibroblasts in immune responses, inflammatory diseases and therapeutic implications. Nat Rev Rheumatol. 2025; 21 (6): 336–54.
17. D'Orazio A, Cirillo AL, Greco G, Ruscio ED, Latorre M, Pisani F, et al. Pathogenesis of rheumatoid arthritis: one year in review 2024. Clin Exp Rheumatol. 2024; 42 (9): 1707–13.
18. Jones DH, Kong YY, Penninger JM. Role of RANKL and RANK in bone loss and arthritis. Ann Rheum Dis. 2002; 61 (Suppl 2): ii32–9.
19. Müller-Ladner U, Pap T, Gay RE, Neidhart M, Gay S. Mechanisms of disease: the molecular and cellular basis of joint destruction in rheumatoid arthritis. Nat Clin Pract Rheumatol. 2005;1(2):102–10.
20. Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev.2010;233(1):233–55.
21. Szekanecz Z, Besenyei T, Paragh G, Koch AE. New insights in synovial angiogenesis. Joint Bone Spine. 2010;77(1):13–9.
22. Klein D. The tumor vascular endothelium as decision maker in cancer therapy. Front Oncol. 2018;8:367.
23. Buckley CD, Pilling D, Lord JM, Akbar AN, Scheel-Toellner D, Salmon M. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol. 2021;22(4):199–204.
24. Filer A, Parsonage G, Smith E, Osborne C, Thomas AMC, Curnow SJ, et al. Differential survival of leukocyte subsets mediated by synovial, bone marrow, and skin fibroblasts: site-specific versus activation-dependent survival of T cells and neutrophils. Arthritis Rheum. 2006;54(7):2096–108.
25. Tang Y, Wang B, Sun X, Li H, Ouyang X, Wei J, et al. Rheumatoid arthritis fibroblast-like synoviocytes co-cultured with PBMC increased peripheral CD4+ CXCR5+ ICOS+ T cell numbers. Clin Exp Immunol. 2017; 190 (3): 384–93.
26. Tran CN, Davis MJ, Tesmer LA, Endres JL, Motyl CD, Smuda C, et al. Presentation of arthritogenic peptide to antigen-specific T cells by fibroblast-like synoviocytes. Arthritis Rheum. 2007;56(5):1497–506.
27. Carmona-Rivera C, Carlucci PM, Moore E, Lingampalli N, Uchtenhagen H, James E, et al. Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci Immunol. 2017;2(10):eaag3358.
28. Reparon-Schuijt CC, van Esch WJ, van Kooten C, Rozier BC, Levarht EW, Breedveld FC, et al. Regulation of synovial B cell survival in rheumatoid arthritis by vascular cell adhesion molecule 1 (CD106) expressed on fibroblast-like synoviocytes. Arthritis Rheum. 2000;43(5):1115–21.
29. Burger JA, Zvaifler NJ, Tsukada N, Firestein GS, Kipps TJ. Fibroblast-like synoviocytes support B-cell pseudoemperipolesis via a stromal cell-derived factor-1- and CD106 (VCAM-1)-dependent mechanism. J Clin Invest. 2001;107(3):305–15.
30. Bombardieri M, Kam N-W, Brentano F, Choi K, Filer A, Kyburz D, et al. A BAFF/APRIL-dependent TLR3-stimulated pathway enhances the capacity of rheumatoid synovial fibroblasts to induce AID expression and Ig class-switching in B cells. Ann Rheum Dis. 2011;70(10):1857–65.
31. Dennis B, Loacker-Schöch K, Classen T, Jose J, Schneider M, Pongratz G. Fibroblast-like synoviocytes preferentially induce terminal differentiation of IgD+ memory B cells instead of naïve B cells. Immunology. 2024;173(3):520–35.
32. Corsiero E, Caliste M, Jagemann L, Fossati-Jimack L, Goldmann K, Cubuk C, et al. Autoimmunity to stromal-derived autoantigens in rheumatoid ectopic germinal centers exacerbates arthritis and affects clinical response. J Clin Invest. 2024;134(12):e169754.
33. Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, et al. Human interleukin-17: a T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999;42 (5):963–70.
34. Take Y, Nakata K, Hashimoto J, Tsuboi H, Nishimoto N, Ochi T, et al. Specifically modified osteopontin in rheumatoid arthritis fibroblast-like synoviocytes supports interaction with B cells and enhances production of interleukin-6. Arthritis Rheum. 2009;60(12):3591–601.
35. Firestein GS. Every joint has a silver lining. Science. 2007;315(5814):952–3.
36. Filer A. The fibroblast as a therapeutic target in rheumatoid arthritis. Curr Opin Pharmacol. 2013;13(3):413–9.
37. Svensson MN, Zoccheddu M, Yang S, Nygaard G, Secchi C, Doody KM, et al. Synoviocyte-targeted therapy synergizes with TNF inhibition in arthritis reversal. Sci Adv. 2020;6(26):eaba4353.
38. Finch R, Sostelly A, Sue-Ling K, Blaeuer A, Duchateau-Nguyen G, Ukarma L, et al. Results of a phase 2 study of RG6125, an anti-cadherin-11 monoclonal antibody, in rheumatoid arthritis patients with an inadequate response to anti-TNFα therapy. Ann Rheum Dis. 2019;78(suppl. 2):189.
39. Takeuchi T, Tanaka Y, Erdman J, Kaneko Y, Saito M, Higashitani C, et al. ASP5094, a humanized monoclonal antibody against integrin alpha-9, did not show efficacy in patients with rheumatoid arthritis refractory to methotrexate: results from a phase 2a, randomized, double-blind, placebo-controlled trial. Arthritis Res Ther. 2020;22(1):252.
40. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506(7488):376–81.
41. Snir O, Gomez-Cabrero D, Montes A, Perez-Pampin E, Gómez-Reino JJ, Seddighzadeh M, et al. Non-HLA genes PTPN22, CDK6 and PADI4 are associated with specific autoantibodies in HLA-defined subgroups of rheumatoid arthritis. Arthritis Res Ther. 2014;16(4):414.
42. Ge X, Frank-Bertonelj M, Klein K, McGovern A, Kuret T, Houtman M, et al. Functional genomics atlas of synovial fibroblasts defining rheumatoid arthritis heritability. Genome Biol 2021;22(1):247.
43. Hosoya T, Saito T, Baba H, Tanaka N, Noda S, Komiya Y, et al. Chondroprotective effects of CDK4/6 inhibition via enhanced ubiquitin-dependent degradation of JUN in synovial fibroblasts. Rheumatology (Oxford). 2022;61(8):3427–38.
44. Buss H, Handschick K, Jurrmann N, Pekkonen P, Beuerlein K, Müller H, et al. Cyclin-dependent kinase 6 phosphorylates NF-κB P65 at serine 536 and contributes to the regulation of inflammatory gene expression. PLoS One. 2012;7(12):e51847.
45. Handschick K, Beuerlein K, Jurida L, Bartkuhn M, Müller H, Soelch J, et al. Cyclin-dependent kinase 6 is a chromatin-bound cofactor for NF-κB-dependent gene expression. Mol Cell. 2014;53(2):193–208.
46. Gao J, Zheng M, Wu X, Zhang H, Su H, Dang Y, et al. CDK inhibitor palbociclib targets STING to alleviate autoinflammation. EMBO Rep. 2022;23(6):e53932.
47. Taniguchi K, Kohsaka H, Inoue N, Terada Y, Ito H, Hirokawa K, et al. Induction of the p16INK4a senescence gene as a new therapeutic strategy for the treatment of rheumatoid arthritis. Nat Med. 1999;5(7):760–7.
48. Nasu K, Kohsaka H, Nonomura Y, Terada Y, Ito H, Hirokawa K, et al. Adenoviral transfer of cyclin-dependent kinase inhibitor genes suppresses collagen-induced arthritis in mice. J Immunol. 2000;165(12):7246–52.
49. Nonomura Y, Nagasaka K, Hagiyama H, Sekine C, Nanki T, Tamamori-Adachi M, et al. Direct modulation of rheumatoid inflammatory mediator expression in retinoblastoma protein-dependent and -independent pathways by cyclin- dependent kinase 4/6. Arthritis Rheum. 2006;54(7):2074–83.
50. Sekine C, Sugihara T, Miyake S, Hirai H, Yoshida M, Miyasaka N, et al. Successful treatment of animal models of rheumatoid arthritis with small-molecule cyclin-dependent kinase inhibitors. J Immunol. 2008;180(3):1954–61.
51. Hosoya T, Iwai H, Yamaguchi Y, Kawahata K, Miyasaka N, Kohsaka H. Cell cycle regulation therapy combined with cytokine blockade enhances antiarthritic effects without increasing immune suppression. Ann Rheum Dis. 2016;75(1):253–9.
52. Andrikopoulou A, Fiste O, Liontos M, Dimopoulos M-A, Zagouri F. Aromatase and CDK4/6 inhibitor-induced musculoskeletal symptoms: a systematic review. Cancers. 2021;13(3):465.
53. Andrikopoulou A, Fiste O, Apostolidou K, Skafida E, Markellos C, Liontos M, et al. CDK4/6 inhibitors and arthralgia: a single institution experience. Med Sci. 2021;9(2):42.
54. Skafida E, Andrikopoulou A, Terpos E, Markellos C, Moustafa S, Pectasides D, et al. Impact of CDK4/6 inhibitors on aromatase inhibitor-associated musculoskeletal syndrome (AIMSS) in the adjuvant setting. Breast J. 2023;2023:3614296.
55. Murakami F, Horimoto Y, Shimizu H, Tada K, Yamaji K, Tamura N, et al. Amelioration of rheumatoid arthritis in a breast cancer patient treated with palbociclib: a case report. Mod Rheumatol. 2021;5(2):214–7.
56. Spring LM, Zangardi ML, Moy B, Bardia A. Clinical management of potential toxicities and drug interactions related to cyclin-dependent kinase 4/6 inhibitors in breast cancer: practical considerations and recommendations. Oncologist. 2017;22(9):1039–48.
57. Guillaume Z, Medioni J, Lillo-Lelouet A, Marret G, Oudard S, Simonaggio A. Severe cellular immunodeficiency triggered by the CDK4/6 inhibitor palbociclib. Clin Breast Cancer. 2020;20(2):e192–5.
58. Bas O, Erul E, Guven DC, Aksoy S. Infectious complications of cyclin-dependent kinases 4 and 6 inhibitors in patients with hormone-receptor-positive metastatic breast cancer: a systematic review and meta-analysis. Support Care Cancer. 2022;30(11):9071–8.
59. Buller W, Pallan L, Chu T, Khoja L. CDK4/6 inhibitors in metastatic breast cancer, a comparison of toxicity and efficacy across agents in a real-world dataset. J Oncol Pharm Practice. 2023;29(8):1825–35.
60. Wekking D, Lambertini M, Dessi M, Denaro N, Bardanzellu F, Garrone O, et al. CDK4/6 inhibitors in the treatment of metastatic breast cancer: focus on toxicity and safety. Semin Oncol. 2023;50(6):131–9.
61. Liu YS, Dong K, Park C. Risk of cardiovascular events with cyclin-dependent kinase 4 and 6 (CDK 4/6) inhibitors among patients with advanced breast cancer: a systematic review and network meta-analysis. Rev Cardiovasc Med. 2023;24(11):309.
62. Nomura J, Tsujimoto S, Tamura K, Yamamoto W, Takahashi H, Horie K, et al. Pharmacological and safety profiles of cyclin-dependent kinase 4/6 inhibitor, candidate for development as rheumatoid arthritis therapeutic option [abstract]. Arthritis Rheum. 2017; 69 (Suppl 10).
63. Tsujimoto S, Horie K, Mashiko T, Nomura J, Kobayashi T. Cyclin-dependent kinase 4/6 inhibitor: a promising development candidate targeting synovial hypertrophy for rheumatoid arthritis treatment [abstract]. Arthritis Rheum. 2018; 70 (Suppl 9).
64. ClinicalTrials.gov. A Study to Assess the Safety and Efficacy of Different Doses of ELV001 to Treat Active Rheumatoid Arthritis in Patients With an Inadequate Response to Methotrexate and Tumor Necrosis Factor Inhibition (START SYNERGY). USA: ClinicalTrials.gov; 2026. Avaliable from: https://clinicaltrials.gov/study/NCT07409103.
65. Genovese MC, Cohen S, Moreland L, Lium D, Robbins S, Newmark R, et al. Combination therapy with etanercept and anakinra in the treatment of patients with rheumatoid arthritis who have been treated successfully with methotrexate. Arthritis Rheum. 2004;50(5):1412–9.