Abstract
Cardiac sarcomeres express a variant of troponin I (cTnI) that contains a unique N-terminal extension of ~30 amino acids with regulatory phosphorylation sites. The extension is important in the control of myofilament response to Ca2+, which contributes to the neuro-humoral regulation of the dynamics of cardiac contraction and relaxation. Hearts of various species including humans express a stress-induced truncated variant of cardiac troponin I (cTnI-ND) missing the first ~30 amino acids and functionally mimicking the phosphorylated state of cTnI. Studies have demonstrated that upregulation of cTnI-ND potentially represents a homeostatic mechanism as well as an adaptive response in pathophysiology including ischemia/reperfusion injury, beta adrenergic maladaptive activation, and aging. We present evidence showing that cTnI-ND can modify the trigger for hypertrophic cardiomyopathy (HCM) by reducing the Ca2+ sensitivity of myofilaments from hearts with an E180G mutation in α-tropomyosin. Induction of this truncation may represent a therapeutic approach to modifying Ca2+-responses in hearts with hypercontractility or heat failure with preserved ejection fraction.
Keywords
Sarcomeres, Ca2+-sensitivity, Hypertrophic cardiomyopathy, Heart failure, Tropomyosin
Highlights
• Evidence shows there is a stress induced proteolysis truncating the N-terminus of cTnI
• N-terminal deleted cardiac TnI promotes adaptive responses to stresses
• The truncated cardiac TnI decreased elevated Ca2+-sensitivity in an HCM model
Abbreviations
cTnI-ND: N-terminal truncated cardiac troponin I; Tm180: α-tropomyosin mutation at E180G; Tm-WT: α-Tropomyosin Wild Type; cTnI-WT: Cardiac Troponin I Wild-Type; cTnI: Cardiac Troponin I; cTn: Cardiac Troponin complex; cTnT: Cardiac Troponin T; cTnC: Cardiac Troponin C; HCM: Hypertrophic Cardiomyopathy
Introduction
In evidence presented here, we discuss a novel adaptive role for physiological and pathological restricted proteolysis of cardiac troponin I (cTnI), which is uniquely expressed in cardiac sarcomeres. cTnI is the inhibitory unit of the heterotrimeric troponin complex (cTn) that controls Ca2+-dependent activation of sarcomeres. Early evidence demonstrated a release of cardiac troponin I (cTnI) into serum in myocardial infarction (MI). This finding led to the development of specific high affinity antibodies for detection of serum cTnI, and led to a vast literature on their use as the preferred marker for MI as well as other cardiac related pathologies (see Solaro and Solaro [1]; Katrukha and Katrukha [2] for recent reviews). Adding to the complexity of its use as a biomarker is evidence that there is a proteolysis of cTnI occurring in the necrosis and necroptosis in these pathologies. Understanding the nature of these products of proteolysis in serum and correlating these data with clinical profiles has been proposed to be important in diagnosis, prognosis, and stratification of patients [1,2].
In contrast to the pathological proteolysis of cTnI, evidence reviewed here has demonstrated an adaptive role for stress-induced, restricted proteolysis of cTnI generating an NH2-deletion (cTnI-ND) of 27-30 amino acids without affecting the core structure in the rest of the molecule [3-5]. As illustrated in Figure 1, cTnI was named for its action as the inhibitory unit of the heterotrimeric Tn complex, which together with cardiac troponin T (cTnT), the tropomyosin (Tm) binding unit, and cardiac troponin C (cTnC), the Ca2+-binding unit, act to modify the of Tm-actin interaction in regulating the on-off state of thin filaments in sarcomere activation and relaxation [6,7]. In diastole, cTnI structures located in an inhibitory peptide (Ip) and at the mobile COOH-domain interact strongly with actin and Tm and promote protein-protein interactions with cTnT-Tm to establish an inhibited state with Tm blocking the reaction of cross-bridges with actins (Figure 1). With the release of Ca2+ into the sarcomere space, Ca2+ binds to the NH2 lobe of cTnC exposing a hydrophobic patch of amino acids that react with a cTnI switch peptide (Sw) thereby releasing the thin filament from its inhibited state. These actions of cTnI are modulated by phosphorylation of cTnI contained in its unique NH2 extension (Figure 1) [8]. The major regulatory phosphorylation sites at S23 and S24 are substrates for protein kinase A [8,9]; a tyrosine kinase phosphorylates cTnI at Tyr 26 [10]. These phosphorylations depress myofilament Ca2+ sensitivity of tension development in detergent extracted (skinned) fiber preparations. Compared to the phosphorylated state, unphosphorylated cTnI NH2 stabilizes the cTnI-SwcTnC activated state by binding relatively tightly to cTnC regulatory site II and the region of Sw binding (Figure 1). When cTnI is phosphorylated, binding of its NH2- terminus with cTnC is modified inducing a weakening of the cTnI-Sw-cTnC interaction [11]. Structure-function and cross-linking studies demonstrated that when cTnI is phosphorylated, the acidic domain in the highly mobile cTnI-NH2-terminus is free to interact in an intramolecular interaction with the basic Ip [12,13]. Moreover, epitope mapping studies have reported that these NH2- terminal interactions induce long range protein-protein interactions modifying the thin filament including regions that interact with cTnT and the Ip [12,14]. The result of this phosphorylation induced weakening of cTnI binding to cTnC is a desensitization of the sarcomeres to the rising Ca2+ in systole and faster relaxation as Ca2+ falls towards the diastolic state. These effects of cTnI phosphorylation during adrenergic stimulation and activation of PKA signaling induce a relaxant effect and abbreviate the cycle time of the heartbeat to tune cardiac function to the prevailing heartbeat [8,15].
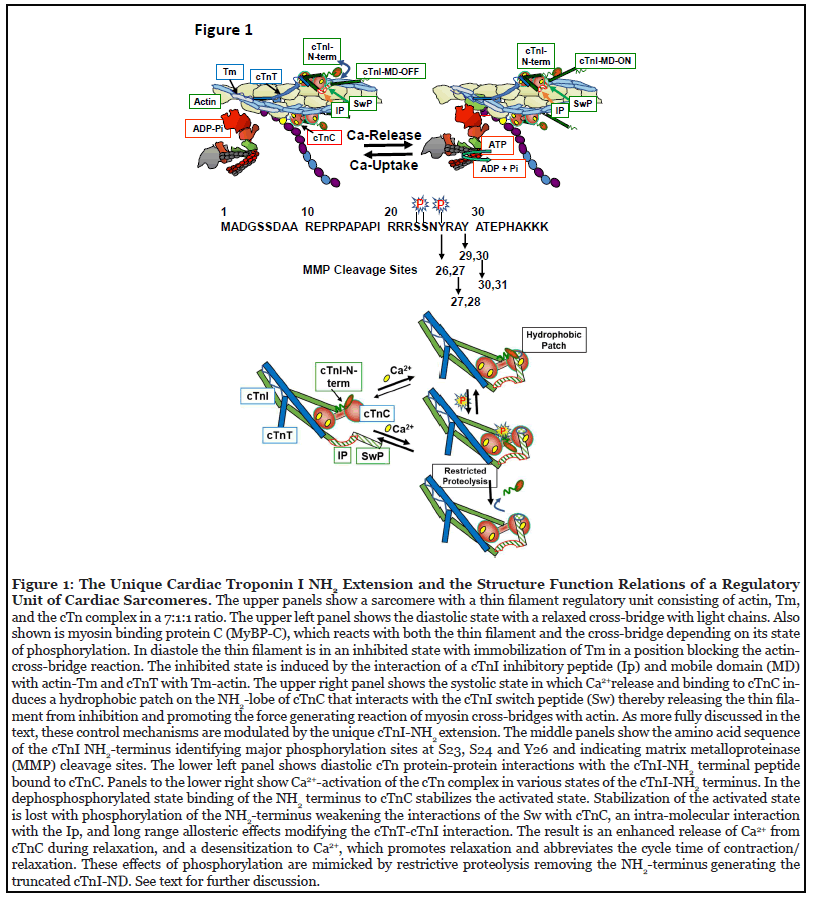
Adaptive Restrictive Proteolysis of Cardiac Troponin I
As expected, truncation of cTnI by restrictive proteolysis removes these effects of interactions at its NH2-terminus with the sarcomere creating a sustained state of sarcomere inhibition with increased cardiac dynamics that mimics the effects of phosphorylation [8,16,17]. Yu et al. [3,4] first reported that a restrictive proteolysis of cTnI removing the NH2-terminal extension occurred in rats subjected to hind limb suspension, simulating micro-gravity. These studies demonstrated that the generation of cTnI-ND did not affect the stoichiometry of the sarcomere proteins, maintained sarcomere Ca2+-response, reduced sarcomere tension generation, and appeared to protect the heart from the elevated neurohumoral signaling and hemodynamic stress in micro-gravity.
Importantly, cTnI-ND was present in the normal sarcomeres in various species including human hearts suggesting a physiological role in modulating contraction and relaxation reserve. Consideration of cTnI-ND as a novel adaptive response was a major conclusion from this early study. There is also evidence that compared to controls myofibrils partially regulated by cTnI-ND show a potential modification of Ca2+-response of the actomyosin ATPase cycle at the level of the rate limiting phosphate release step controlling relaxation [23]. In comparison to control unloaded isolated ventricular cardiac myocytes, cTnI-ND expressing myocytes had shorter resting lengths whereas the diameter and resting sarcomere length were unchanged. With activation there was an increase in the amplitude of contraction and faster shortening and relengthening, which are non-additive with the effect of isoproterenol treatment [26]. Moreover, there was an increase in early rates of fall of the Ca2+ transient with no change in peak amplitude or basal levels suggesting an enhanced release of Ca2+ from cTnC during relaxation [18,26]. Transgenic mice expressing cTnI-ND in the cardiac compartment confirmed its role in modulating diastolic function in the intact heart [17].
The discovery that cTnI-ND is normally present and functional in myofibrils indicates a potential to modulate response to β-adrenergic stimulation. Interestingly the relative amounts of cTnI-ND expressed are low in cardiac myocytes with relatively high prevailing β-adrenergic stimulation and PKA dependent phosphorylation and higher in myocytes with reduced PKA dependent phosphorylation [18]. An important question is the proteolytic process generating cTnI-ND. Likely candidates are mu-calpain and matrix metalloproteinases (MMPs) both of which have been identified to be present in association with cardiac myofibrils. Mu-calpains have been reported to be important in the proteolytic NH2 degradation of cTnT [19] and MyBP-C [20]. Mu-calpains are also responsible for degradation of cTnI, but have not been specifically identified in the adaptive generation of cTnI-ND. There is evidence that treatment of cTn with MMP cleaves at multiple locations including those in the generation of cTnI-ND in animal models (Figure 1) [21]. MMPs have been shown to be upregulated in HCM [22], but there have been no studies of whether early increases in MMP activity represent an adaptive process.
Our hypothesis is that as with many control mechanisms there is a homeostatic range of proteolytic activity that is adaptive and elevated activities outside this range are maladaptive.
Restricted Proteolysis of cTnI and Cardiac Remodeling
Evidence has been developed indicating the potential significance of the generation of cTnI-ND in chronic physiological and pathological remodeling of the myocardium. Results of our studies comparing aging related modifications in cardiac and sarcomere function in controls and Tg-cTnI-ND mice emphasize a role of restricted proteolysis of cTnI in physiological adaptation [23]. Intra-ventricular pressure measurements in hearts at 4 months of age showed control mice already had depressed diastolic function compared to Tg-cTnI-ND hearts. Measurement of LV end-diastolic pressure that was relatively high and -dp/dt that were relatively low in the 4-month controls were significantly improved in the 4-month old Tg-cTnI-ND hearts. Working heart preparations showed a prolonged diastolic duration and abbreviated systolic duration in Tg-cTnI-ND compared to controls. Investigations of the associated changes in sarcomere Ca2+-response together with measurements of ATP hydrolysis showed that compared to controls skinned fibers from hearts of cTnI-ND hearts were less sensitive to Ca2+, with a steeper response to Ca2+, and an increase in cross-bridge kinetics as determined from the tension cost (ratio of unit ATP hydrolyzed/unit of tension) [24]. Importantly skinned fibers from controls and Tg-cTnI-ND mice had no differences in the population of the alphamyosin heavy chain (α-MHC) with fast kinetics and the slow β-MHC with slow kinetics. Echocardiography studies comparing hearts of these mice beating in situ at 16 months of age reported significant diastolic abnormalities in ejection fraction, cardiac output, and intra-ventricular relaxation times in the controls which were prevented in the Tg-cTnI-ND mice [24].
There are also studies indicating a role for the generation of cTnI-ND in chronic remodeling in cardiac disorders. Compared to controls, myocytes from human hearts in failure commonly demonstrate diastolic abnormalities associated with low levels of cTnI phosphorylation at the PKA sites and increased myofilament response to Ca2+ [25]. Thus, it seemed reasonable to speculate that under these conditions the presence of cTnI-ND would be beneficial in restoring relaxation kinetics. In a study interrogating the possible up regulation of cTnI-ND in a heart failure model induced by β-adrenergic signaling deficiency (Gsα-DF) developed by a conditional cardiac specific deficiency of Gsα, Feng et al. [5] demonstrated an increase in expression of cTnI-ND from 10% of total cTnI in controls to 30% in the Gsα-DF mouse hearts. To show that this restricted proteolysis was adaptive, Feng et al. [5] crossed the Gsα- DF mice with mice expressing cardiac cTnI-ND. Double transgenic mice were protected from the progression of heart failure occurring in the single transgenic Gsα-DF mice. An application of sarcomere inhibition by cTnIND is in familial hypertrophic cardiomyopathy (HCM) in which there is hyper-contractility mainly associated with mutations of sarcomere proteins inducing an increase in myofilament Ca2+-sensitivity thought to be the biophysical trigger for the clinical course of the disorder. In the case of the diastolic abnormality associated with expression of a mutant cTnI (cTnI-R193H) linked to restrictive cardiomyopathy (RCM), Li et al. [26] reported that crossbreeding Tg-cTnI-ND mice with the Tg-cTnI-R193H mice was able to attenuate the effect of cTnI-R193H, reduce the increased Ca2+-response, and partially restore diastolic function close to control levels. Although these data demonstrate a minimization of diastolic abnormalities in hearts of TG-cTnI-R193H when crossed with cTnIND [26], the mutant cTnI-R193H was partially replaced by cTnI-ND. Thus, it was difficult to know whether the improvement in function was due directly to a general desensitization of the myofilaments. Along these lines, we tested whether sustained increased sarcomere inhibition and relaxation associated with transgenic expression of a phospho-mimetic cTnI-S23D, S24D (cTnI-DD) was able to modify disease progression in an HCM mouse model (Tg- αTmE180G) expressing a mutant of another sarcomere protein other than a cTnI mutant, which increased Ca2+- response [27]. We crossed Tg-cTnI-DD mice with Tg-αTm- E180G mice and demonstrated a significant improvement in diastolic dysfunction as well as significantly reduced fibrosis [28]. This has led us to test whether cTn-ND can modify the pathologic trigger associated with a maladaptive response in the myofilaments regulated by αTm-E180G (Tm-180).
cTnI-ND Reduces the Biophysical Trigger in Sarcomeres Expressing a Mutant Tm Linked to HCM
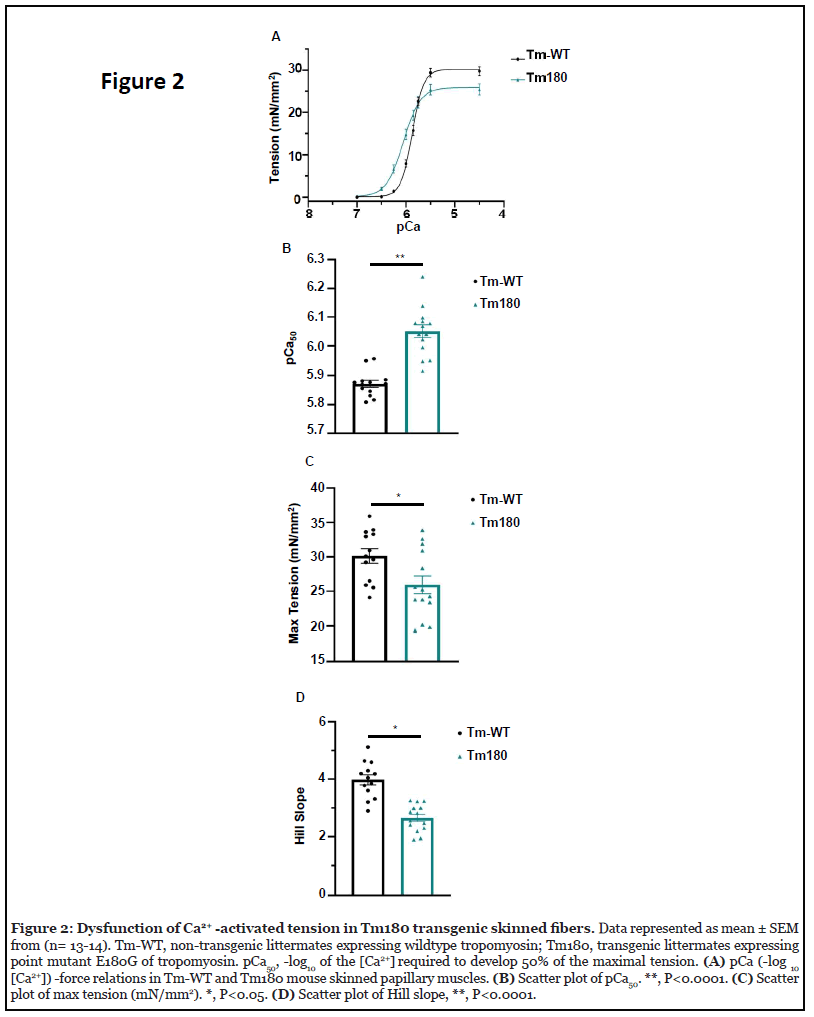
To test the ability of cTnI-ND to reduce the increased response to Ca2+ in myofilaments controlled by mutant sarcomere proteins other than mutant cTnI, we employed detergent-extracted (skinned) fiber bundles dissected from papillary muscles of WT mice and transgenic mice expressing the HCM linked mutation α-Tm-E180G (Tm-180) [27]. Materials and methods used in these experiments are detailed in the supplement. Figure 2 shows data confirming the increased response to Ca2+ in skinned fibers isolated from control hearts and hearts expressing Tm-180. Figure 2A illustrates the relation between free Ca2+ expressed as pCa (-log [Ca]) and steady-state tension developed by WT fibers and Tm-180 fibers. The left shift of the pCa-tension relation of the Tm-180 fibers compared to WT indicates an increase in Ca2+ -response considered the trigger for HCM. Figure 2B demonstrates this significant increase in Ca2+ -response of the Tm-180 fibers showing that half-maximal tension occurred at a higher pCa50 (pCa at half-maximum tension) i.e. lower free Ca2+ concentration compared to the WT fibers. Data in Figure 2C shows that Tm-180 fibers developed a lower maximum tension than the the WT fibers. To illustrate the relative steepness of the pCa-tension relations, Figure 2D shows the value of Hill coefficients (Hill Slope), a measure of cooperative activation of the myofilaments. Tm-180 fibers show less cooperative activation than the WT fibers (Figure 2D).
For incorporating WT cTnI and cTnI-ND into the Tm- 180 myofilaments, we generated WT cTnI and cTnIND in a bacterial expression system and reconstituted troponin complexes containing either wild-type cTnI-WT or cTnI-ND, expressed Myc-labeled cardiac troponin T (cTnT) as a marker, and expressed TnC-WT. As previously demonstrated [28], addition of an excess of these reconstituted complexes to skinned fibers under optimal conditions induces an exchange of the exogenous complex with native cTn. We determined the exchange efficiency by expression of the myc-tagged cTnT during the production of the exogenous cTn complex, which allowed us to separate the exogenous vs. endogenous troponin based on molecular weight. Importantly there was no significant difference in exchange efficiency between the Tm180 fibers exchanged with cTnI-ND vs. Tm180 fiber exchanged with cTnI-WT containing Tn complex (Figure 3A, 3B).
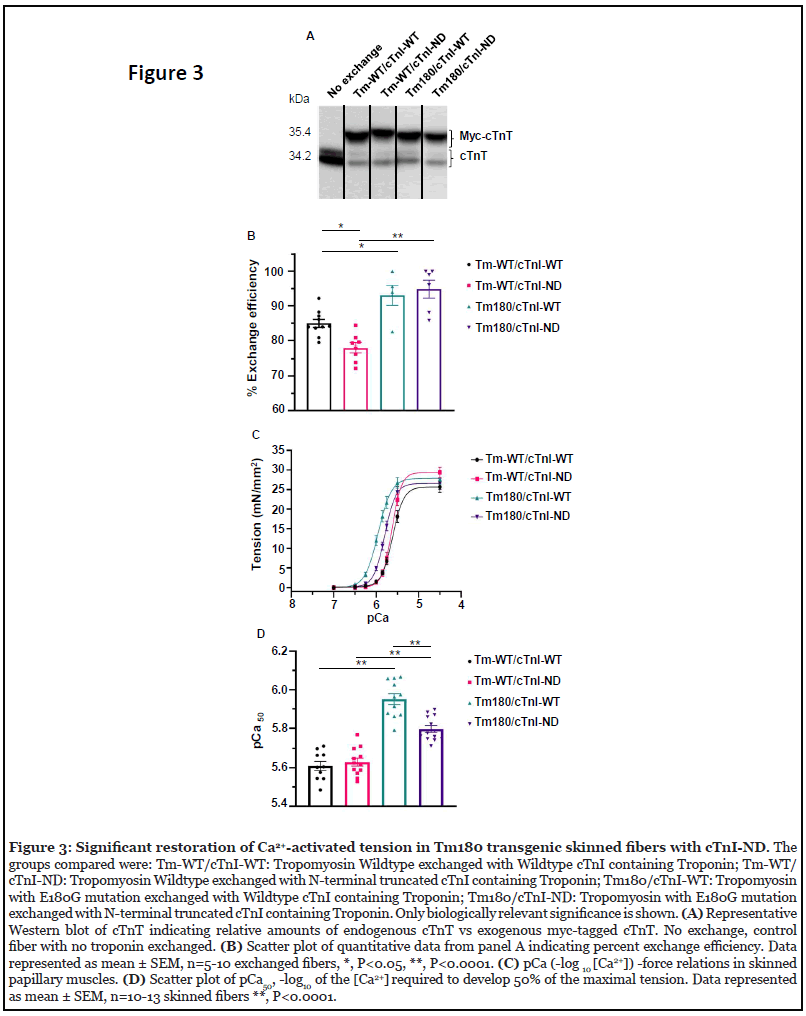
The exchange efficiency for the Tm-WT fiber exchanged with cTnI-ND was about 78% whereas the Tm-WT fiber exchanged with cTnI-WT was significantly higher at 85% (Figure 3A, 3B). Tm-WT vs Tm180 exchanged with cTnIWT was significantly increased by 8% (Figure 3A, 3B). Furthermore, Tm-WT vs Tm180 exchanged with cTnI-ND was also significantly increased by 17% (Figure 3A, 3B). However, there were no significant differences between Tm180 fibers exchanged with either cTnI-WT or cTnI-ND, which was important to analyze the effects of Ca2+ sensitivity on the background of this mutation (Figure 3A, 3B). In the data reported here, Tm-WT skinned fibers controlled by either cTnI-WT or cTnI-ND showed no differences in the pCa-tension relation (Figure 3C, 3D, Table 1). In contrast to this finding with an exchange of cTnI-WT with cTnIND, our previous studies in skinned fibers from hearts of TG-cTnI-ND mice [24] demonstrated a desensitization compared to controls. This difference suggests that other modifications may occur in situ. One possibility is the limited expression of α-smooth muscle actin (α-SMA) in myofibrils, which has been reported to occur in young TGcTnI- ND mouse hearts [29], which was not present in the current study. There are also possible post-translational modifications occurring in vivo in the transgenic hearts. Nevertheless our objective was to show that the presence of cTnI-ND can alter the biophysical triggering of HCM signaled by sarcomere protein mutations.
| Groups | Max Tension (mN/mm2) | Hill Slope (nH) | pCa50 | n = skinned fibers |
|---|---|---|---|---|
| Tm-WT/cTnI-WT | 25.68 ± 1.36 | 3.50 ± 0.15 | 5.61 ± 0.02∧ | 10 |
| Tm-WT/cTnI-ND | 29.35 ± 1.26 | 4.30 ± 0.22 *,# | 5.63 ± 0.02$ | 12 |
| Tm180/cTnI-WT | 27.88 ± 1.33 | 2.99 ± 0.10 | 5.95 ± 0.03† | 11 |
| Tm180/cTnI-ND | 26.55 ± 1.41 | 3.38 ± 0.07 | 5.80 ± 0.02 | 13 |
Table 1: Ca2+ -activated tension parameters for exchanged fibers.
As expected, the Ca2+ sensitivity was significantly increased in the Tm180 fiber exchanged with cTnI-WT vs Tm-WT fibers exchanged with cTnI-WT (Figure 3C, 3D, Table 1), recapitulating the results found with non-exchanged fibers (Figure 2) and further validating the exchange fiber approach [30-32]. The Tm180 fibers exchanged with cTnI-ND vs. cTnI-WT showed a significant decrease in the myofilament’s response to Ca2+ (Figure 3C, 3D, Table 1), indicating the presence of cTnI-ND in the Tn complex is sufficient to partially restore myofilament Ca2+ sensitivity in the etiology of a myofilament mutation disease. The effects of cTnI-ND and cTnI-S23D, S24D to correcte myofilament response to Ca2+ inform the identifications of targets for small molecules for use in HCM. Targeting the cTnI-cTnC interface with such compounds is also aided by earlier work [12,13,33] and recent findings [11] identifying the regions of interaction of the unphosphorylated NH2 region of cTnI with the cTnI switch-peptide binding region as well as the Ca2+ binding site II of cTnC.
Conclusions and Future Directions
Ample evidence indicates a novel mechanism of control of cardiac contractility by a restrictive proteolysis of cTnI removing its unique regulatory NH2 terminal extension. These findings also support the idea that a general mechanism of control of cardiac sarcomeres is restrictive proteolysis, which has been reported in proteolytic degradation of cTnT [20]. Studies in aging-induced and cardiac disorder-induced diastolic abnormalities indicate the potential for therapeutic applications of our findings. The effects of cTnI-ND provide information for targeting the sarcomere with small molecules or peptides such as that derived the COOH-terminus of cTnI [34]. The success in the use of small molecules for inhibition of the hypercontractility in the clinical course of advanced HCM [35] indicates the importance of detection of new targets to address the diverse responses to triggering mutations in HCM [36]. Approaches that identify mechanisms for specific activation of proteolytic cascades to generate cTnI-ND also provide an intriguing challenge to future investigations.
Acknowledgments
This work was supported by NIH grants RO1 HL128468 (to BMW and RJS), PO1 HL062426 Project 1 and Core C (to RJS, BMW, and CMW), RO1 HL127691 (to JPJ and RJS).
Author Contributions
CMW: Writing-Original Draft, Writing- Review & Editing, Methodology, Investigation; MH: Data Collection and Analysis, Writing- Review & Editing; HZF: Resources, Writing- Review & Editing; BMW: Writing- Review & Editing Funding acquisition; JPJ: Conceptualized, Funding acquisition, Writing- Review & Editing; RJS: Writing-Original Draft, Writing- Review & Editing, Conceptualized, Funding acquisition.
Declaration of Interests
R. John Solaro is a member of the Scientific Advisory Board of Cytokinetics, Inc., a consultant to Pfizer, Inc. and Myokardia/Bristol Myers Squibb. The authors declare no additional competing financial interests.
References
2. Katrukha IA, Katrukha AG. Myocardial Injury and the Release of Troponins I and T in the Blood of Patients. Clin Chem. 2021;67(1):124-30.
3. Yu ZB, Bao JX, Ma J, Zhang LF, Jin JP. Changes in myocardial contractility and contractile proteins after four weeks of simulated [correction of simulate] weightlessness in rats. J Gravit Physiol. 2000;7(2):P147-8.
4. Yu ZB, Zhang LF, Jin JP. A proteolytic NH2-terminal truncation of cardiac troponin I that is up-regulated in simulated microgravity. J Biol Chem. 2001;276(19):15753-60.
5. Feng HZ, Chen M, Weinstein LS, Jin JP. Removal of the N-terminal extension of cardiac troponin I as a functional compensation for impaired myocardial beta-adrenergic signaling. J Biol Chem. 2008;283(48):33384-93.
6. Solís C, Solaro RJ. Novel insights into sarcomere regulatory systems control of cardiac thin filament activation. J Gen Physiol. 2021;153(7).
7. Sheng JJ, Jin JP. TNNI1, TNNI2 and TNNI3: Evolution, regulation, and protein structure-function relationships. Gene. 2016;576(1 Pt 3):385-94.
8. Solaro RJ, Henze M, Kobayashi T. Integration of troponin I phosphorylation with cardiac regulatory networks. Circ Res. 2013;112(2):355-66.
9. Kobayashi T, Solaro RJ. Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu Rev Physiol. 2005;67:39-67.
10. Salhi HE, Walton SD, Hassel NC, Brundage EA, de Tombe PP, Janssen PM, et al. Cardiac troponin I tyrosine 26 phosphorylation decreases myofilament Ca2+ sensitivity and accelerates deactivation. J Mol Cell Cardiol. 2014;76:257-64.
11. Kachooei E, Cordina NM, Potluri PR, Guse JA, McCamey D, Brown LJ. Phosphorylation of Troponin I finely controls the positioning of Troponin for the optimal regulation of cardiac muscle contraction. J Mol Cell Cardiol. 2020;150:44-53.
12. Howarth JW, Meller J, Solaro RJ, Trewhella J, Rosevear PR. Phosphorylation-dependent conformational transition of the cardiac specific N-extension of troponin I in cardiac troponin. J Mol Biol. 2007;373(3):706-22.
13. Warren CM, Kobayashi T, Solaro RJ. Sites of intra- and intermolecular cross-linking of the N-terminal extension of troponin I in human cardiac whole troponin complex. J Biol Chem. 2009;284(21):14258-66.
14. Akhter S, Jin JP. Distinct conformational and functional effects of two adjacent pathogenic mutations in cardiac troponin I at the interface with troponin T. FEBS Open Bio. 2015;5:64-75.
15. Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, et al. Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ Res. 2001;88(10):1059-65.
16. Solaro RJ, Moir AJ, Perry SV. Phosphorylation of troponin I and the inotropic effect of adrenaline in the perfused rabbit heart. Nature. 1976;262(5569):615-7.
17. Barbato JC, Huang QQ, Hossain MM, Bond M, Jin JP. Proteolytic N-terminal truncation of cardiac troponin I enhances ventricular diastolic function. J Biol Chem. 2005;280(8):6602-9.
18. McConnell BK, Popovic Z, Mal N, Lee K, Bautista J, Forudi F, et al. Disruption of protein kinase A interaction with A-kinase-anchoring proteins in the heart in vivo: effects on cardiac contractility, protein kinase A phosphorylation, and troponin I proteolysis. J Biol Chem. 2009;284(3):1583-92.
19. Zhang Z, Biesiadecki BJ, Jin JP. Selective deletion of the NH2-terminal variable region of cardiac troponin T in ischemia reperfusion by myofibril-associated mu-calpain cleavage. Biochemistry. 2006;45(38):11681-94.
20. Barefield DY, McNamara JW, Lynch TL, Kuster DWD, Govindan S, Haar L, et al. Ablation of the calpain-targeted site in cardiac myosin binding protein-C is cardioprotective during ischemia-reperfusion injury. J Mol Cell Cardiol. 2019;129:236-46.
21. Mahmud Z, Zahran S, Liu PB, Reiz B, Chan BYH, Roczkowsky A, et al. Structure and proteolytic susceptibility of the inhibitory C-terminal tail of cardiac troponin I. Biochim Biophys Acta Gen Subj. 2019;1863(4):661-71.
22. Bi X, Yang C, Song Y, Yuan J, Cui J, Hu F, et al. Matrix Metalloproteinases Increase Because of Hypoperfusion in Obstructive Hypertrophic Cardiomyopathy. Ann Thorac Surg. 2021;111(3):915-22.
23. Gunther LK, Feng HZ, Wei H, Raupp J, Jin JP, Sakamoto T. Effect of N-Terminal Extension of Cardiac Troponin I on the Ca(2+) Regulation of ATP Binding and ADP Dissociation of Myosin II in Native Cardiac Myofibrils. Biochemistry. 2016;55(12):1887-97.
24. Biesiadecki BJ, Tachampa K, Yuan C, Jin JP, de Tombe PP, Solaro RJ. Removal of the cardiac troponin I N-terminal extension improves cardiac function in aged mice. J Biol Chem. 2010;285(25):19688-98.
25. van der Velden J, Papp Z, Zaremba R, Boontje NM, de Jong JW, Owen VJ, et al. Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res. 2003;57(1):37-47.
26. Li Y, Charles PY, Nan C, Pinto JR, Wang Y, Liang J, et al. Correcting diastolic dysfunction by Ca2+ desensitizing troponin in a transgenic mouse model of restrictive cardiomyopathy. J Mol Cell Cardiol. 2010;49(3):402-11.
27. Prabhakar R, Boivin GP, Grupp IL, Hoit B, Arteaga G, Solaro RJ, et al. A familial hypertrophic cardiomyopathy alpha-tropomyosin mutation causes severe cardiac hypertrophy and death in mice. J Mol Cell Cardiol. 2001;33(10):1815-28.
28. Alves ML, Dias FAL, Gaffin RD, Simon JN, Montminy EM, Biesiadecki BJ, et al. Desensitization of myofilaments to Ca2+ as a therapeutic target for hypertrophic cardiomyopathy with mutations in thin filament proteins. Circ Cardiovasc Genet. 2014;7(2):132-43.
29. Kern S, Feng HZ, Wei H, Cala S, Jin JP. Up-regulation of alpha-smooth muscle actin in cardiomyocytes from nonhypertrophic and non-failing transgenic mouse hearts expressing N-terminal truncated cardiac troponin I. FEBS Open Bio. 2013;4:11-7.
30. Chandra M, Kim JJ, Solaro RJ. An improved method for exchanging troponin subunits in detergent skinned rat cardiac fiber bundles. Biochem Biophys Res Commun. 1999;263(1):219-23.
31. Sumandea MP, Pyle WG, Kobayashi T, de Tombe PP, Solaro RJ. Identification of a functionally critical protein kinase C phosphorylation residue of cardiac troponin T. J Biol Chem. 2003;278(37):35135-44.
32. Warren CM, Karam CN, Wolska BM, Kobayashi T, de Tombe PP, Arteaga GM, et al. Green Tea Catechin Normalizes the Enhanced Ca2+ Sensitivity of Myofilaments Regulated by a Hypertrophic Cardiomyopathy-Associated Mutation in Human Cardiac Troponin I (K206I). Circulation Cardiovascular genetics. 2015;8(6):765-73.
33. Cheng Y, Lindert S, Kekenes-Huskey P, Rao VS, Solaro RJ, Rosevear PR, et al. Computational studies of the effect of the S23D/S24D troponin I mutation on cardiac troponin structural dynamics. Biophys J. 2014;107(7):1675-85.
34. Hornos F, Feng HZ, Rizzuti B, Palomino-Schatzlein M, Wieczorek D, Neira JL, et al. The muscle-relaxing C-terminal peptide from troponin I populates a nascent helix, facilitating binding to tropomyosin with a potent therapeutic effect. J Biol Chem. 2021;296:100228.
35. Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;351(6273):617-21.
36. Neubauer S, Kolm P, Ho CY, Kwong RY, Desai MY, Dolman SF, et al. Distinct Subgroups in Hypertrophic Cardiomyopathy in the NHLBI HCM Registry. J Am Coll Cardiol. 2019;74(19):2333-45.