Abstract
Junctophilin-2 (JP2) is a key structural protein of junctional membrane complexes (JMCs) that stabilize contacts between the sarcoplasmic reticulum and transverse tubules required for excitation-contraction (EC) coupling in cardiomyocytes. Under pathophysiological conditions, the intracellular cysteine protease Calpain activated by disturbed intracellular Ca2+ homeostasis cleaves JP2 and, hence, disturbs EC coupling. In return, the primary N-terminal JP2 cleavage fragment (NT1) translocates into the nucleus, where it accumulates in dense local spots associated with gene-rich euchromatin and proposedly acts as a cardioprotective transcriptional regulator in heart failure. Secondary cleavage events by Calpain or other proteases may further alter the function of JP2. After recently revealing the complete spectrum of JP2 cleavage fragments generated by Calpain proteolysis, we provide here an update about the position of the Calpain cleavage sites relative to the structural environment of JP2 in JMCs and how heart disease-associated gene variants of JP2 could affect the proteolytic regulation by Calpain.
Keywords
Calcium, Calpain, Cardiomyocyte, Junctophilin-2, Ryanodine Receptor
Introduction
Calpains are Ca2+-activated cysteine proteinases cleaving specific substrates in Ca2+ signaling pathways involved in a wide variety of biological functions. Among 15 known Calpain isoforms, Calpain-1 and Calpain-2 are most common and ubiquitously expressed in mammalian cells [1,2]. Calpain activation has been reported to be involved in various neurodegenerative [3], neuromuscular [4], and cardiac disorders [5,6]. The development and testing of Calpain inhibitors as therapeutic agents is the subject of intense ongoing research [3,5,7]. One emerging target of Calpain cleavage under pathophysiological conditions is Junctophilin-2 (JP2) [8,9]. Recently, we systematically analyzed the JP2 cleavage products as a function of Calpain-1 versus Calpain-2 proteolytic activities [10]. Together with newly available structural data for JP2 [11], our work provides new insights into the proteolytic regulation of JP2 by Calpain relevant for biomedical research as described below.
Calpain-Specific Cleavage Sites of JP2 and Its Structural Environment
Full-length JP2 is an important structural protein stabilizing junctional membrane complexes (JMC) between the sarcoplasmic reticulum (SR) and Transverse (T-) tubules, i.e., plasma membrane (PM) invaginations in cardiomyocytes [12,13]. JMCs couple the action potential propagation along the PM and T-tubules with the release of the secondary messenger Ca2+ from the SR lumen that serves as Ca2+ stores. In striated muscles, membrane depolarization-induced Ca2+ release mediates a cascade of signaling events that leads to cell contraction, i.e., a process known as excitation-contraction (EC) coupling [13]. Under disease conditions, a disturbed intracellular Ca2+ homeostasis can lead to persistent, pathologic activation of Calpain cleaving JP2 and, hence, disrupting JMCs. In return, loss of JMCs and maladaptive ultrastructural remodeling of T-tubules causes abnormal EC (un)coupling that contributes to impaired cardiac performance, rhythm disorders, or muscle weakness [9,14].
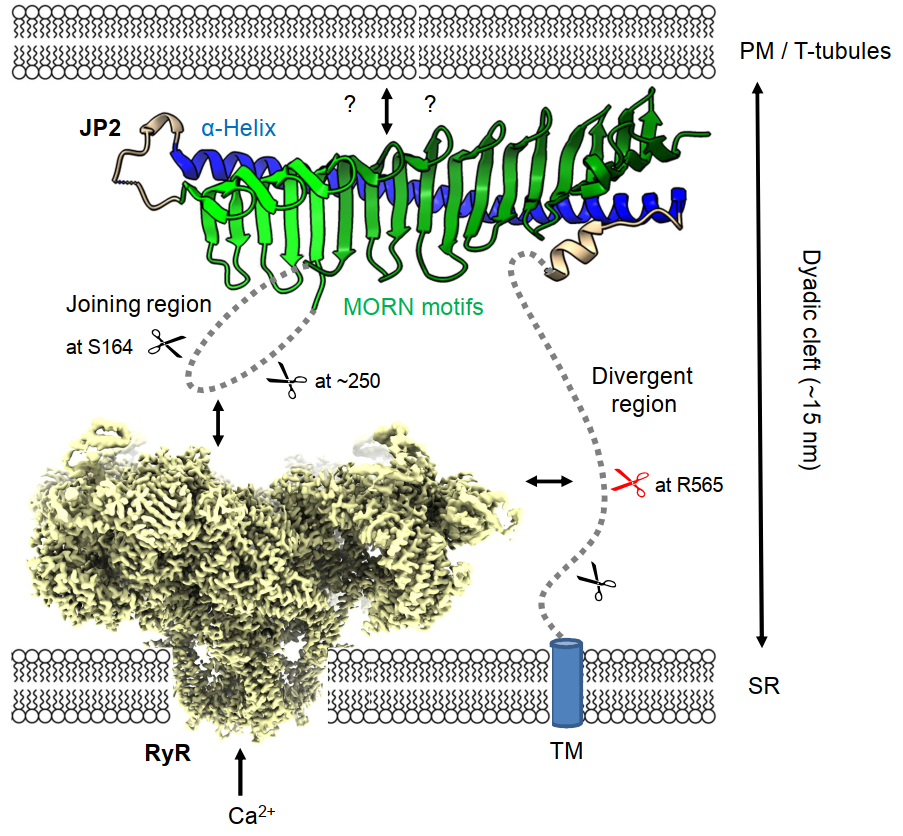
To stabilize JMCs, JP2 interacts with the PM directly or indirectly through binding to voltage-dependent L-type Ca2+ channels (LTCC) and SR membrane at its N- and C-terminus, respectively (Figure 1) [11]. I.e., JP2 possesses eight highly conserved N-terminal Membrane Occupation and Recognition Nexus (MORN) motifs that were traditionally thought to mediate binding to certain phospholipid head groups at the inner leaflet surface of PM/T-tubules and a C-terminal transmembrane domain (TM) tail-anchors JP2 into the SR membrane [12,15,16]. Interestingly, the recently reported crystal structure of an N-terminal JP2 fragment shows the MORN motifs formed as β-hairpins that are included in a 19-stranded antiparallel β-sheet [11]. The β-sheet has a twisted shape similar to a cradle being concave at the inner and convex at the outer surface. The convex surface is bound to a long α-helix (αH) forming a stabilizing backbone along the β-sheet cradle (Figure 1). The N-terminal concave surface acts as a binding site for a C-terminal peptide of rabbit CaV1.1 (a.a. 1594-1609) [11]. CaV1.1 (skeletal isoform) and CaV1.2 (cardiac isoform) are pore-forming α1 subunits of LTCC that function as voltage sensors in T-tubules [17,18]. JP2-LTCC interactions may thus contribute to indirect T-tubule binding of JMCs through LTCCs and efficient nanodomain EC coupling.
The joining region, which is a long flexible linker between MORN1-6 and MORN7-8, and the divergent region connecting αH and TM are unresolved in the JP2 crystal structure [11] and AlphaFold2 predictions (UniProt protein ID: Q9BR39) [19,20], presumably because of higher intrinsic disorder [10,11]. In our recent publication, we showed that the joining region and divergent region of JP2 represent hot spots for Calpain cleavage sites using in vitro calpain cleavage assays of purified proteins [10]. Calpain preferentially cleaves mouse JP2 in the divergent region at residue R565. Subsequently, three secondary Calpain cleavage events occur, i.e., two in the joining region at residues S164 and ~250 and one in the divergent region near the TM domain (Figure 1). Increased protein instability by, inter alia, Calpain digestion has been associated with the presence of PEST motifs, which are local protein sequence regions rich in proline (P), glutamate (E), aspartate, and serine (S) or threonine (T) residues [21-23]. Indeed, the primary and secondary Calpain cleavage sites of mouse JP2 are in close proximity to predicted PEST sequence signatures, two in the joining region and three in the divergent region. Interestingly, the PEST motif with the highest score (a.a. 565-589, score: 26.85) overlaps with the primary Calpain cleavage site of JP2 at R565 [10]. PEST sequences are frequently characterized by higher protein backbone flexibility [24], which may enable a better steric accessibility and broad conformation spectrum for diverse interactions of the joining region and divergent region.
Since αH forms a backbone stabilizing the MORN motifs, the nanometric (~15 nm) dyadic cleft between PM and SR is supposedly bridged by the divergent region instead of αH as previously proposed in most JP2 topology models. This would place the divergent region in an exposed central position and make it easily accessible to Calpain digestion if not protected by protein interactions. Interestingly, both the joining region [25] and divergent region [26] have been reported to interact with the Ryanodine Receptor (RyR), a huge (>2.2 MDa) tetrameric SR Ca2+ release channel in JMCs [27-29]. These JP2-RyR interactions may protect JP2 against Calpain cleavage by covering the Calpain cleavage sites (Figure 1). We showed that knockout of RyR2 (cardiac isoform) in 2-month-old human iPSC-derived cardiomyocytes destabilizes full-length JP2 resulting in an increase of the Calpain-specific primary cleavage fragments, while RyR2 appeared to be more resistant to Calpain cleavage than JP2 in cardiomyocyte lysates treated with Calpain-1 or -2 [10]. Hence, RyR2 seems to shield JP2 against Calpain-dependent cleavage presumably through protein-protein interactions.
Genetic JP2 Variants
LTCC binding and interactions with other JMC components supposedly support JP2-RyR2 interactions enriched in higher local concentrations in JMCs, while JP2 variants may alter calpain cleavage and disrupt protective RyR2 or LTCC binding. JP2 genetic variations have been mainly associated with hypertrophic cardiomyopathy [25,30-35], however, 82 variants localize directly to the recently crystalized MORN-αH domains [11] and more than 140 variants to the full-length JP2 protein (Figure 2). As little functional characterization exists for JP2 variants particularly the large number of putative missense mutations, the structural prediction of pathogenic or misfolded products provides avenues for new mechanistic rationales. I.e. first, 6 JP2 variants (G21E, W64*, G117C, Y129D, G136S, G142*) directly alter highly conserved consensus MORN residues, predicted to abolish their stabilizing interactions [11]. Second, replacement of JP2 glycine residues that contribute to αH binding may destabilize the correct β-sheet folding of the MORN repeats [11]. Third, the backbone helix αH contains alanine residues organized in a highly conserved heptad repeat pattern aligning with the β-sheet’s glycine valley such that substitution by 16 variants, likely missense mutations, can create steric hindrance or destabilize binding of the backbone helix αH [11].
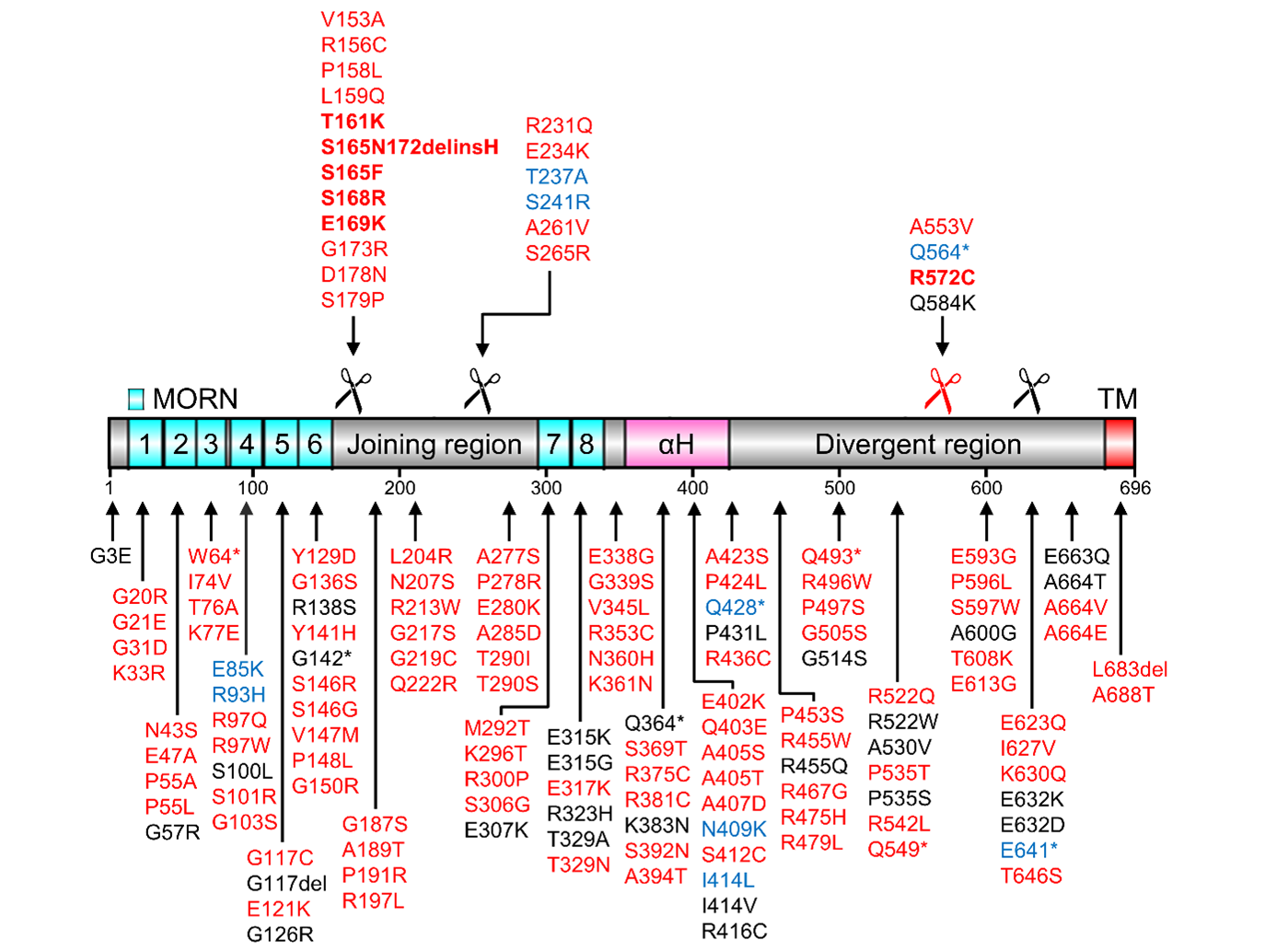
Moreover, a number of JP2 variants may affect substrate recognition by Calpain or prevent cleavage. Of special interest are JP2 variants that are located in the joining region or divergent region close to Calpain-specific cleavage sites (Figure 2). A direct JP2 variant of the primary Calpain cleavage site conserved in human JP2 at R572 is R572C [13]. Importantly, R572C is a Arg-to-Cys missense mutation of the scissile bond in the primary cleavage site (569-YAVR↓TTPP-576). According to Calpain cleavage predictions, arginine is a preferred residue on the N-terminal side of the scissile bond (P1) while the mutation to cysteine renders the P1 position disfavored for Calpain cleavage [23,36]. Hence, the R572C variant is predicted to be a less suitable Calpain substrate than WT JP2. Accordingly, the computational tools GPS-CCD (v1.0) [37] and LabCaS [38] determined substantially lower cleavage score values for R572C (GPS-CCD score: 0.75; LabCaS score: 0.0026) than for WT JP2 (GPS-CCD score: 1.02; LabCaS score: 0.0044) (Figure 3A). The R572C variant has been linked to hypertrophic cardiomyopathy but is otherwise undescribed and effects of R572C on Calpain cleavage have not yet been reported. The same is true for several other hypertrophic cardiomyopathy-linked JP2 variants in the divergent region near R572, e.g., R522Q, P535T, R542L, A553V, E593G, P596L, S597W, T608K, E613G, E623Q, I627V, K630Q [13]. JP2 variants that introduce a stop codon at Q549 or at Q564 preventing synthesis of full-length JP2 (incl. C-terminal TM) have also been linked to hypertrophic or dilated cardiomyopathy, respectively [13].
In the joining region of JP2, several genetic variants associated with hypertrophic or dilated cardiomyopathy are clustered near the secondary Calpain cleavage site of JP2 at S164 (e.g., V153A, R156C, P158L, L159Q, T161A, S165F, S165N172delinsH, S168R, E169K, G173R) and at ~250 (e.g., R231Q, E234K, T237A, S241R, A261V, S265R) [10,11,13]. The Calpain cleavage site of mouse JP2 at S164 is conserved in human at S164 (161-TSLS↓SLRS-168) [10]. In the S165F variant, the C-terminal side of the scissile bond (P1’) is mutated from serine to phenylalanine substituting a favored for a disfavored residue in P1’ [23,36]. Indeed, GPS-CCD [37] and LabCaS [38] calculate substantially lower cleavage scores for S165F compared to WT JP2 (Figure 3B). The same applies to the S165N172delinsH variant, in which a.a. 165-172 are replaced by one histidine residue (Figure 3B). Both variants are linked to hypertrophic cardiomyopathy [30,39]. S165F mutation of JP2 has been reported to abolish phosphorylation by protein kinase C (PKC) and reduce EC coupling in skeletal myotubes [40]. If the S164F variant or the putative PKC phosphorylation of JP2 at S165 affects secondary Calpain cleavage events at S164 or ~250 is yet unknown. The Calpain cleavage of JP2 at S164 is directly overlapping with three more genetic variants, i.e., T161A (in P4), S168R (in P4’), and E169K (in P5'), indicating high genetic variation around S164. E169K has been implicated in atrial fibrillation pathogenesis due to reduced binding of JP2 to RyR2 [25]. Thus, the region around the Calpain cleavage site S164 seems to be directly involved in JP2-RyR2 interactions.
Hence, genetic variations may affect the proteolytic regulation of JP2 by Calpain due to altered cleavage site preferences or steric accessibilities. In return, altered JP2 stability or altered cleavage fragments could moderate a different response to Calpain activation in heart failure.

Nuclear Translocation of NT1
Calpain proteolytic regulation alters the function of JP2. After the loss of TM due to Calpain cleavage of JP2 at R565, the N-terminal fragment (NT1) translocates into the nucleus, where it is proposed to function as a cardio-protective transcriptional regulator in heart failure [10,41]. We observed increased intranuclear NT1 in isolated mouse cardiomyocytes treated with Ca2+/Ionomyocin and in hiPSC-CMs after RyR2-KO [10]. Hence, the lipid-protein and protein-protein interactions of NT1 in the JMC environment are not strong enough to keep NT1 associated with T-tubules (incl. LTCC) or SR (incl. RyR2) once the C-terminal TM domain is cleaved off. As supposedly responsible for the nuclear translocation, two highly conserved nuclear localization signals (NLS) have been discovered in the sequence of mouse JP2, namely a bipartite NLS (a.a. 345-359) located directly in front of αH [41] and a monopartite NLS (a.a. 484-492) in the divergent region [42]. Thus, NT1 (JP21-565) retains both NLS after primary Calpain cleavage. Interestingly, Lahiri et al. reported nuclear translocation of an alternative C-terminal JP2 fragment after Calpain-2 specific cleavage of mouse JP2 at residue G482 [42]. Accordingly, the C-terminal JP2 cleavage product (CTP) would contain the monopartite NLS (a.a. 484-492). Since blocking of CTP nuclear translocation protected cardiomyocytes from developing isoproterenol-induced hypertrophy in vitro, Lahiri et al. proposed a maladaptive role of CTP in heart failure development contrary to the cardio-protective of NT1 [42].
However, we could not confirm Calpain-2 cleavage of JP2 at G482 or nuclear translocation of a C-terminal JP2 fragment. I.e. first, deletion mutation Δ479-486 did not alter the cleavage pattern of purified JP2 by Calpain-1 or -2 compared to WT, whereas Δ563-568 completely abolished the primary Calpain cleavage event for both isoforms [10]. Second, our confocal and STED (Stimulated Emission Depletion) imaging of Ca2+/Ionomycin-treated cardiomyocytes and hiPSC-CM RyR2-KO did not detect intranuclear signals after immunostaining against a C-terminal JP2 epitope (JP2 antibody H-3, sc-377086, Santa Cruz) but immunodetected NT1 (JP2 antibody 70R-6923, Fitzgerald). Similarly, Wang et al. could only detect primary Calpain cleavage of JP2 at R565 but not at G482 using in vitro calpain cleavage assays of recombinant HEK293 overexpression lysates [43]. Contrary to our findings, they reported higher proteolytic efficiencies for Calpain-1 compared to Calpain-2 in cleaving JP2 and other cardiac substrates (incl. Troponin T, Troponin I, and β2-spectrin) overexpressed in HEK293 cells [43]. Different recombinant overexpression levels of Calpain-1 vs Calpain-2 in the crude cell lysates may explain the discrepancy in the apparent proteolytic efficiencies. In untreated 2-month-old hiPSC-CM, we observed a closer association between JP2 and Calpain-2 compared to Calpain-1 [10]. Future studies are needed to explore in detail the (patho-)physiological activation conditions and Calpain isoforms responsible for JP2 cleavage at which cleavage site(s).
Strikingly, our work demonstrates that intranuclear NT1 organizes in small dense accumulation spots, spatially associated with transcriptionally active euchromatic regions, where NT1 is proposed to bind through an alanine-rich region DNA binding motif to TATA box motifs to repress heart disease-reactive MEF2-dependent gene expression [10,41]. Transcriptionally permissive euchromatin is often segregated from transcriptionally repressed heterochromatin, giving rise to the A and B compartments, respectively [44,45]. Euchromatin is further organized into transcriptionally active pockets interspersed by inactive domains [46,47]. As a potential mechanism for the exclusion of inactive chromatin from the active compartment high levels of RNA and RNA-binding proteins in the active nuclear compartment has been proposed [48]. Moreover, within the active compartment and favoring the selective retention of active chromatin, transcribed genes often associate with assemblies of RNA polymerase II, transcription factors, and RNA transcripts [49-51]. Recently, small-scale microphases have been demonstrated that form the typical pattern of euchromatin organization, where RNA remains tethered to transcribed euchromatin through RNA polymerases [52]. Hence, while it remains to be fully established at the euchromatin level, it seems plausible that NT1 accumulates in separate small transcriptionally active pockets to repress reactive MEF2-dependent genes.
Conclusion
JP2 is an important structural protein of JMCs required for efficient EC coupling [13]. The Calpain proteolytic regulation alters the function of JP2 in cardiomyocytes. After the loss of the C-terminal TM domain, the primary N-terminal cleavage fragment NT1 translocates into the nucleus, where it accumulates in dense local spots associated with gene-rich euchromatin and proposedly acts as a cardioprotective transcriptional regulator in heart failure [10,41]. JP2 interactions with RyR2 and its structural environment in intact JMCs seem to be protective against Calpain cleavage [10]. Genetic variants of JP2 can weaken these protective interactions making JP2 easier accessible to Calpain or directly alter the cleavage site preferences in JP2. Alteration in JP2 stability or in JP2 proteolytic fragmentation could moderate a different response to Calpain activation in heart failure. After recently revealing the complete spectrum of JP2 cleavage fragments generated by Calpain proteolysis [10], we assigned here for the first time genetic JP2 variants directly to Calpain cleavage sites. In particular, R572C and S165F are predicted to impair the Calpain-specific primary cleavage site of JP2 at R572 and the secondary cleavage site at S164, respectively. On the other hand, E169K proposedly weakens protective JP2-RyR2 interactions [25] without directly interfering with Calpain cleavage prediction scores (Figure 3). Future studies are wanted to elucidate if these hypertrophic cardiomyopathy-linked variants have an impact on the proteolytic regulation of JP2 by Calpain.
Acknowledgments
This project was funded by Deutsche Forschungsgemeinschaft (DFG) through Collaborative Research Unit SFB1190 to SEL (project P03) and was supported under Germany’s Excellence Strategy (EXC2067/1-390729940).
Author Contributions
Design of research: G.W., S.E.L.; Data analysis and interpretation of results: G.W., S.E.L.; Prepared figures and drafted manuscript: G.W., S.E.L.; Edited and revised manuscript: G.W., S.E.L. All authors have read and approved the final manuscript.
Competing Interests
The authors declare no competing interests.
References
2. Ono Y, Sorimachi H. Calpains: an elaborate proteolytic system. Biochim Biophys Acta. 2012;1824:224-36.
3. Camins A, Verdaguer E, Folch J, Pallas M. Involvement of calpain activation in neurodegenerative processes. CNS Drug Rev. 2006;12:135-48.
4. Ozyilmaz B, Kirbiyik O, Ozdemir TR, Ozer OK, Kutbay YB, Erdogan KM, et al. Experiences in the molecular genetic and histopathological evaluation of calpainopathies. Neurogenetics. 2022;23:103-14.
5. Wang Y, Chen B, Huang CK, Guo A, Wu J, Zhang X, et al. Targeting Calpain for Heart Failure Therapy: Implications From Multiple Murine Models. JACC Basic Transl Sci. 2018;3:503-17.
6. Letavernier E, Zafrani L, Perez J, Letavernier B, Haymann JP, Baud L. The role of calpains in myocardial remodelling and heart failure. Cardiovasc Res. 2012;96:38-45.
7. Ono Y, Saido TC, Sorimachi H. Calpain research for drug discovery: challenges and potential. Nat Rev Drug Discov. 2016;15:854-76.
8. Murphy RM, Dutka TL, Horvath D, Bell JR, Delbridge LM, Lamb GD. Ca2+-dependent proteolysis of junctophilin-1 and junctophilin-2 in skeletal and cardiac muscle. J Physiol. 2013;591:719-29.
9. Wu CY, Chen B, Jiang YP, Jia Z, Martin DW, Liu S, et al. Calpain-dependent cleavage of junctophilin-2 and T-tubule remodeling in a mouse model of reversible heart failure. J Am Heart Assoc. 2014;3:e000527.
10. Weninger G, Pochechueva T, El Chami D, Luo X, Kohl T, Brandenburg S, et al. Calpain cleavage of Junctophilin-2 generates a spectrum of calcium-dependent cleavage products and DNA-rich NT1-fragment domains in cardiomyocytes. Sci Rep. 2022;12:10387.
11. Yang ZF, Panwar P, McFarlane CR, Tuinte WE, Campiglio M, Van Petegem F. Structures of the junctophilin/voltage-gated calcium channel interface reveal hot spot for cardiomyopathy mutations. Proc Natl Acad Sci U S A. 2022;119:e2120416119.
12. Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6:11-22.
13. Lehnart SE, Wehrens XHT. The Role of Junctophilin Proteins in Cellular Function. Physiol Rev. 2022;102(3):1211-1261.
14. van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, Gaur N, et al. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation. 2011;123:979-88.
15. Beavers DL, Landstrom AP, Chiang DY, Wehrens XH. Emerging roles of junctophilin-2 in the heart and implications for cardiac diseases. Cardiovasc Res. 2014;103:198-205.
16. Brandenburg S, Pawlowitz J, Eikenbusch B, Peper J, Kohl T, Mitronova GY, et al. Junctophilin-2 expression rescues atrial dysfunction through polyadic junctional membrane complex biogenesis. JCI Insight. 2019;4.
17. Wolf M, Eberhart A, Glossmann H, Striessnig J, Grigorieff N. Visualization of the domain structure of an L-type Ca2+ channel using electron cryo-microscopy. J Mol Biol. 2003;332:171-82.
18. Wu J, Yan Z, Li Z, Yan C, Lu S, Dong M, et al. Structure of the voltage-gated calcium channel Cav1.1 complex. Science. 2015;350:aad2395.
19. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583-9.
20. Perni S. The Builders of the Junction: Roles of Junctophilin1 and Junctophilin2 in the Assembly of the Sarcoplasmic Reticulum-Plasma Membrane Junctions in Striated Muscle. Biomolecules. 2022;12(1):109.
21. Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986;234:364-8.
22. Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci. 1996;21:267-71.
23. Tompa P, Buzder-Lantos P, Tantos A, Farkas A, Szilagyi A, Banoczi Z, et al. On the sequential determinants of calpain cleavage. J Biol Chem. 2004;279:20775-85.
24. Sandhu KS, Dash D. Conformational flexibility may explain multiple cellular roles of PEST motifs. Proteins. 2006;63:727-32.
25. Beavers DL, Wang W, Ather S, Voigt N, Garbino A, Dixit SS, et al. Mutation E169K in junctophilin-2 causes atrial fibrillation due to impaired RyR2 stabilization. J Am Coll Cardiol. 2013;62:2010-9.
26. Luo T, Yan N, Xu M, Dong F, Liang Q, Xing Y, et al. Junctophilin-2 allosterically interacts with ryanodine receptor type 2 to regulate calcium release units in mouse cardiomyocytes. Gen Physiol Biophys. 2022;41:133-40.
27. Marks AR, Tempst P, Hwang KS, Taubman MB, Inui M, Chadwick C, et al. Molecular cloning and characterization of the ryanodine receptor/junctional channel complex cDNA from skeletal muscle sarcoplasmic reticulum. Proc Natl Acad Sci U S A. 1989;86:8683-7.
28. Zalk R, Clarke OB, des Georges A, Grassucci RA, Reiken S, Mancia F, et al. Structure of a mammalian ryanodine receptor. Nature. 2015;517:44-9.
29. Miotto MC, Weninger G, Dridi H, Yuan Q, Liu Y, Wronska A, et al. Structural analyses of human ryanodine receptor type 2 channels reveal the mechanisms for sudden cardiac death and treatment. Sci Adv. 2022;8:eabo1272.
30. Landstrom AP, Weisleder N, Batalden KB, Bos JM, Tester DJ, Ommen SR, et al. Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. J Mol Cell Cardiol. 2007;42:1026-35.
31. Matsushita Y, Furukawa T, Kasanuki H, Nishibatake M, Kurihara Y, Ikeda A, et al. Mutation of junctophilin type 2 associated with hypertrophic cardiomyopathy. J Hum Genet. 2007;52:543-8.
32. Seidelmann SB, Smith E, Subrahmanyan L, Dykas D, Abou Ziki MD, Azari B, et al. Application of Whole Exome Sequencing in the Clinical Diagnosis and Management of Inherited Cardiovascular Diseases in Adults. Circ Cardiovasc Genet. 2017;10.
33. Jones EG, Mazaheri N, Maroofian R, Zamani M, Seifi T, Sedaghat A, et al. Analysis of enriched rare variants in JPH2-encoded junctophilin-2 among Greater Middle Eastern individuals reveals a novel homozygous variant associated with neonatal dilated cardiomyopathy. Sci Rep. 2019;9:9038.
34. Quick AP, Landstrom AP, Wang Q, Beavers DL, Reynolds JO, Barreto-Torres G, et al. Novel junctophilin-2 mutation A405S is associated with basal septal hypertrophy and diastolic dysfunction. JACC Basic Transl Sci. 2017;2:56-67.
35. De Bruijn S, Galloo X, De Keulenaer G, Prihadi EA, Brands C, Helbert M. A special case of hypertrophic cardiomyopathy with a differential diagnosis of isolated cardiac amyloidosis or junctophilin type 2 associated cardiomyopathy. Acta Clin Belg. 2021;76:136-43.
36. Shinkai-Ouchi F, Koyama S, Ono Y, Hata S, Ojima K, Shindo M, et al. Predictions of Cleavability of Calpain Proteolysis by Quantitative Structure-Activity Relationship Analysis Using Newly Determined Cleavage Sites and Catalytic Efficiencies of an Oligopeptide Array. Mol Cell Proteomics. 2016;15:1262-80.
37. Liu Z, Cao J, Gao X, Ma Q, Ren J, Xue Y. GPS-CCD: a novel computational program for the prediction of calpain cleavage sites. PLoS One. 2011;6:e19001.
38. Fan YX, Zhang Y, Shen HB. LabCaS: labeling calpain substrate cleavage sites from amino acid sequence using conditional random fields. Proteins. 2013;81:622-34.
39. Nykamp K, Anderson M, Powers M, Garcia J, Herrera B, Ho YY, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017;19:1105-17.
40. Woo JS, Hwang JH, Ko JK, Weisleder N, Kim DH, Ma J, et al. S165F mutation of junctophilin 2 affects Ca2+ signalling in skeletal muscle. Biochem J. 2010;427:125-34.
41. Guo A, Wang Y, Chen B, Wang Y, Yuan J, Zhang L, et al. E-C coupling structural protein junctophilin-2 encodes a stress-adaptive transcription regulator. Science. 2018;362.
42. Lahiri SK, Quick AP, Samson-Couterie B, Hulsurkar M, Elzenaar I, van Oort RJ, et al. Nuclear localization of a novel calpain-2 mediated junctophilin-2 C-terminal cleavage peptide promotes cardiomyocyte remodeling. Basic Res Cardiol. 2020;115:49.
43. Wang J, Ciampa G, Zheng D, Shi Q, Chen B, Abel ED, et al. Calpain-2 specifically cleaves Junctophilin-2 at the same site as Calpain-1 but with less efficacy. Biochem J. 2021;478:3539-53.
44. Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289-93.
45. Kempfer R, Pombo A. Methods for mapping 3D chromosome architecture. Nat Rev Genet. 2020;21:207-26.
46. Verschure PJ, van Der Kraan I, Manders EM, van Driel R. Spatial relationship between transcription sites and chromosome territories. J Cell Biol. 1999;147:13-24.
47. Iborra FJ, Pombo A, Jackson DA, Cook PR. Active RNA polymerases are localized within discrete transcription "factories' in human nuclei. J Cell Sci. 1996;109(Pt 6):1427-36.
48. Cremer T, Cremer M, Hubner B, Strickfaden H, Smeets D, Popken J, et al. The 4D nucleome: Evidence for a dynamic nuclear landscape based on co-aligned active and inactive nuclear compartments. FEBS Lett. 2015;589:2931-43.
49. Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, et al. Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell. 2018;174:744-57 e24.
50. Cho WK, Spille JH, Hecht M, Lee C, Li C, Grube V, et al. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science. 2018;361:412-5.
51. Wei M, Fan X, Ding M, Li R, Shao S, Hou Y, et al. Nuclear actin regulates inducible transcription by enhancing RNA polymerase II clustering. Sci Adv. 2020;6:eaay6515.
52. Hilbert L, Sato Y, Kuznetsova K, Bianucci T, Kimura H, Julicher F, et al. Transcription organizes euchromatin via microphase separation. Nat Commun. 2021;12:1360.