Abstract
Protease-activated receptors (PARs) and the neurokinin 1 receptor (NK1R) belong to the G protein-coupled receptor (GPCR) family. In this review, we focus on the regulatory mechanism of ectodomain shedding by ADAM10/17 metalloprotease via GPCR signaling. PAR2 and NK1R induce membrane blebbing, resulting in phosphatidylserine externalization in the cellular membrane, which is required for ADAM10/17 metalloprotease activation. Membrane-embedded dual oxidase 2 (DUOX2) has NADPH oxidase and peroxidase domains. NADPH oxidase domain generates hydrogen peroxide (H2O2), while the peroxidase domain produces peroxynitrite (ONOO−) through the interaction of nitrogen oxide with superoxide. Both H2O2 and peroxynitrite activate ADAM10/17 metalloproteases. PAR2 signaling activates ADAM10/17 by NADPH-mediated H2O2, leading to the transactivation of DUOX2/EGFR/TLR4 to synergistically upregulate IL-12p40 production after exposure to LPS. In contrast, nitric oxide (NO) synthesis is promoted by NK1R signaling, and DUOX2 generates ONOO-, preferentially activating ADAM10/17 metalloprotease. Ectodomain shedding of membrane-bound fractalkine is mediated by ADAM10/17. Substance P (SP)/NK1R signals enhance shedding of membrane-bound fractalkine, whereas small interfering RNA for DUOX2 further increases membrane-bound fractalkine but decreases soluble fractalkine compared with cells treated with SP alone. Considering the signaling pathway of TGFβ1 (an inhibitor of iNOS mRNA expression), silencing of RNA for TAK-1 upregulates membrane-bound fractalkine tripartite motif 28 (TRIM28)/transcriptional intermediary factor 1β (TIF1β) functions as an E3 ubiquitin ligase and specificity protein 1 negatively regulate TGFβ1 levels to upregulate the generation of peroxynitrite, leading to increased shedding of membrane-bound fractalkine via SP/NK1R signaling. DUOX2 plays a pivotal role for ectodomain shedding through ADAM10/17 activation by GPCR signaling.
Keywords
GPCR, DUOX2, ADAM family, Peroxynitrite, Shedding
Abbreviations
ADAM: A Disintegrin and Metalloprotease; DUOX: Dual Oxidase; EGFR: Epidermal Growth Factor Receptor; ELISA: Enzyme-linked Immunosorbent Assay; GPCR: G Protein-coupled Receptor; HNE: Human Neutrophil Elastase; iNOS: Inducible Nitric Oxide Synthase; NADPH: Nicotinamide Adenine Dinucleotide Phosphate; NOS2: Nitric Oxide Synthase 2; PAR: Protease-activated Receptor; SP: Substance P; TGF: Tumor Growth Factor; TLR: Toll-like Receptor; TRIM: Tripartite Motif 28; TIF: Transcriptional Intermediary Factor
Introduction
G protein-coupled receptors (GPCRs) are a large family of cell membrane receptors. Neutrophils are the first immune cells to be recruited to inflamed tissue and play a role in the innate immune response. Neutrophils release serine proteases such as neutrophil elastase, proteinase 3, cathepsin G. These proteolytic enzymes activate protease-activated receptors (PARs) to induce intracellular signaling. PARs belong to a family of GPCRs that are activated by proteolytic cleavage of the amino-terminus, and these receptors act as sensors for extracellular proteases. Neutrophils are potent immune effectors against bacterial infections. Macrophages also are important in infections as effectors and regulators [1]. We previously reported that granulocyte macrophage colony-stimulating factor (GM-CSF) upregulates PAR2 expression by human macrophages [2]. Therefore, neutrophils may collaborate with macrophages to modulate immune response via PAR2 signaling. The proinflammatory effects of lipopolysaccharide (LPS) are amplified during pre-existing neutrophilic inflammation, as neutrophils augment LPS-mediated proinflammatory signaling [3]. Neutrophil elastase (NE) is one of the innate effector molecules that modulate immune responses, and plasma NE levels are elevated in patients with pneumonia [ 4]. NE is a major contributor to bacterial infectionassociated host tissue inflammation and damage [5], as it modulates the inflammatory cytokine expression in response to pathogens [6]. Importantly, mice lacking NE were reported to reveal impaired host defense against gram-negative bacterial sepsis [7]. NE activates proteaseactivated receptor 2 (PAR2). Interestingly, PAR2 was shown to interact with TLR4 and enhance TLR4-dependent signaling [8]. NE activates membrane blebbing formation from endothelial cells in a time-dependent manner [9], with membrane blebbing accompanied by phosphatidylserine externalization from the inner plasma membrane leaflet [ 10]. Most importantly, phosphatidylserine exposure has been reported to be required for shedding activation by sheddase—a disintegrin and metalloprotease (ADAM) [ 11,12]. One role of ADAM in epidermal growth factor (EGF) ligand shedding mediated by GPCRs is EGFR transactivation [13,14]. Previously, we reported that human neutrophil elastase (HNE) enhances IL-12p40 production via transactivation of the PAR2/EGFR/TLR4 signaling pathway [15]. In this review, we focus on the role of PAR2 on transactivation of TLR4 signaling by ADAM10/17.
Neurokinin 1 receptor (NK1R) also belongs to GPCR family. Substance P (SP) activates NK1R, and, interestingly, SP/NK1R signaling has been reported to induce membrane blebbing [16]. This led us to hypothesize that SP promotes the shedding activity of ADAM10/17 by phosphatidylserine externalization. The shedding activity of the ADAM family may be closely associated with the development of autoimmune diseases. Autoimmune diseases also are known to be associated with elevated serum levels of neuropeptide SP [17]. The chemokine fractalkine is have shown that serum levels of soluble fractalkine are elevated in inflammatory and autoimmune diseases [18]. Fractalkine shedding is mediated by the sheddase (ADAM) [19]. This makes us further hypothesize that SP/NK1R signaling is closely associated with fractalkine sheddin synthesized as a membrane-bound protein, but studiesg to enhance soluble fractalkine levels. We recently reported that SP induces the shedding of membrane-bound fractalkine in human macrophages [20]. We also review the regulatory mechanism of activation of ADAMs by SP/ NK 1R signaling.
Materials and Methods
Ethics statement
The Board of Ethics in Kumamoto Health Science University approved to obtain blood from volunteers in conformity with the declaration of Helsinki after obtaining their informed consent (No. 17046).
Chemicals and reagents
Human neutrophil elastase (HNE) with an activity of 200 U/L was purchased from SERVA Electrophoresis (Heidelberg, Germany). Recombinant human GM-CSF was purchased from Tocris Bioscience (Bristol, UK). U73122 (Tocris Bioscience) and Rottlerin (Tocris Bioscience) were employed to investigate the intracellular signaling pathways involved in IL-12p40 production. Two proteaseactivated receptor (PAR)-2 agonists (AC- 264613 [2-oxo-4- phenylpyrrolidine-3-carboxylic acid [1-(3-bromo-phenyl)- (E/Z)-ethylidene]-hydrazide], Tocris Bioscience, Bristol, UK and 2-furoyl-LIGRLO-amide, Tocris Bioscience) were purchased to study the intracellular signal transduction pathways involved in PAR2 stimulation. Substance P (Peptide Institute Inc., Osaka, Japan) was employed to investigate the intracellular signaling pathways involved in fractalkine production. N(ω)-nitro-L-arginine methyl ester (L-NAME) also was obtained from Abcam, Cambridge, UK.
Induction of GM-CSF-dependent human macrophages
Peripheral blood mononuclear cells (PBMCs) was obtained from heparinized blood samples [21]. PBMCs collected using Lymphoprep gradients (Axis-Shield PoC As, Norway) were suspended with Lymphocyte medium for thawing (BBLYMPH1, Zen-Bio, Inc. Research Triangle Park, NC). The monocytes were stained with CD14- phycoerythrin (PE) mouse anti-human monoclonal antibody (Life technologies, Staley Road Grand Island, NY). The purity of monocytes was determined by Fluorescence Activated Cell Sorting (FACS), showing 86.6 ± 1.7 % (mean ± SE, n=120, 85.2-89.7). GM-CSF dependent macrophages were obtained after monocytes stimulated with recombinant human GM-CSF on days 1, 3, and 6 of culture [22]. Macrophages (on day 9 of culture) were utilized as GM-CSF dependent macrophages in this study.
Preparation of whole-cell lysates from cell culture
Human macrophages (on day 9 of culture) were stimulated with HNE (5 μM) or SP (5 μM) for 6 hours and culture medium was carefully removed. Mammalian protein extraction reagent (100 μL; M-PER, Thermo Fisher Scientific Inc., Waltham, MA) was pipetted into each well, after which the culture plate was gently shaken for 5 minutes. The lysate was collected and transferred to a microcentrifuge tube for centrifugation at 12,000 g for 10 minutes. The supernatants were used as a whole-cell lysate in this study.
ELISA for IL-12p40, fractalkine, NOS2/iNOS, phosphatidylserine and nitrotyrosine
Macrophages were stimulated with SP (5 μM) for 6 hours. ELISA kits were used to measure levels of IL-12p40 (Abcam, Cambridge, UK), fractalkine (Abcam), NOS2/ iNOS (Abcam), phosphatidylserine (Biocompare, South San Francisco, CA;) and nitrotyrosine protein (Abcam) in whole-cell lysates. The sensitivity of ELISA for the various proteins was as follows: IL-12p40, 20 pg/mL; fractalkine, 0.25 ng/mL; NOS2/iNOS, 18 pg/mL; phosphatidylserine, 0.78 ng/mL; and nitrotyrosine, 50 nM. RNA interferences with DUOX2, ERK1/2, p22phox, PAR2, TRAF6, TLR4, EGFR, TGFβ1/2/3, TAK-1, β-arrestin 2, GRK2, Sp1, C/ EBPβ, TRIM28/TIF1β, or Fli-1 siRNA.
Transfection of macrophages with siRNAs for DUOX2 (50 nM), ERK1/2 (50 nM), p22phox (50 nM), PAR2 (50 nM), TRAF6 (50 nM), TLR4 (50 nM), EGFR (50 nM), TGFβ1/2/3 (50 nM), TAK-1 (50 nM), β-arrestin 2 (50 nM), GRK2 (50 nM), C/EBPβ (50 nM), Sp1 (50 nM), TRIM28/ TIF1β (50 nM), Fli-1 (50 nM) or control siRNA-A (Santa Cruz Biotechnology, Santa Cruz, CA) was performed on day 7-8 of cell culture using Lipofectamine (Life Technologies, Carlsbad, CA). IL-12p40 and fractalkine protein levels in whole-cell lysates or cell-culture supernatants were measured by ELISA.
Western blotting for PAR2, fractalkine, and TGFβ1
The levels of PAR2 in monocytes and macrophages (on day 9 of culture) in whole-cell lysates were detected by western blotting. Furthermore, human macrophages (on day 9 of culture) were exposed to SP (5 μM) for 6 hours, and the levels of fractalkine and TGFβ1 in whole-cell lysates were measured by western blotting. The proteins in the wholecell lysates were separated by sodium dodecyl sulfatepolyacrylamide gel electrophoresis (ATTO corporation, Tokyo, Japan) and transferred onto polyvinylidene fluoride membranes (Thermo Fisher Scientific) for immunoblotting. The membranes were incubated with 0.2 × 103 μg/L mouse anti-human PAR2, fractalkine, and TGFβ1 (Santa Cruz Biotechnology, Santa Cruz, CA) for 1 hour at room temperature, washed, and incubated with alkaline phosphatase-conjugated anti-mouse IgG (Santa Cruz Biotechnology) diluted to 1:5000. Then the membranes were incubated with chemiluminescence enhancer (Immun-Star, Bio-Rad Laboratories, Hercules, CA) and exposed to XAR film (Kodak, Rochester, NY). After the film was developed, bands were quantified with a densitometer and ImageQuant software (Molecular Dynamics, Sunnydale, CA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was also detected by western blotting with an anti-GAPDH antibody (Santa Cruz Biotechnology) and PAR2, fractalkine, and TGFβ1 protein levels were normalized to GAPDH.
Analysis of mRNA expression by reverse transcription quantitative polymerase chain reaction
TGFβ1 mRNA expression was determined by reverse transcription quantitative polymerase chain reaction (RTqPCR). Transfection of macrophages with siRNAs for C/ EBPβ (50 nM), Sp1 (50 nM), TRIM28/TIF1β (50 nM), Fli- 1 (50 nM) or control siRNA-A (Santa Cruz Biotechnology, Santa Cruz, CA) was performed on day 7-8 of cell culture using Lipofectamine (Life Technologies, Carlsbad, CA). On day 9 of culture, macrophages were stimulated with substance P (5 μM) for 6 hours. Total RNA was extracted with Isogen (Nippon Gene, Tokyo, Japan), and equal amounts of RNA were reverse transcribed to obtain cDNA by using a PrimeScript RT kit (TaKaRa Bio Inc., Shiga, Japan). The primer sequences for real-time RT-qPCR were as follows:
5’-TCCTGGCGATACCTCAGCAA-3’ (forward for TGFβ1), 5’-GCTAAGGCGAAAGCCCTCAA-3’ (reverse for TGFβ1), 5’-CTTCGCTCTCTGCTCCTCCTGTTCG-3’ (forward for GAPDH), and 5’-ACCAGGCGCCCAATACGACCAAAT-3’ (reverse for GAPDH). These primers and TB Green realtime PCR master mix were obtained from Takara Bio Inc. (Shiga, Japan). RT-qPCR was performed by using a LightCycler® (Nippon Genetics Co. Ltd., Tokyo, Japan) according to the manufacturer’s instructions, and the level of TGFβ1 mRNA was normalized to that of GAPDH. TGFβ1 mRNA levels were determined by RT-qPCR.
Statistical analysis
Results are expressed as the mean (SE). Differences between two groups were analyzed using a t-test for independent means, and differences between more than two groups were compared by analysis of variance. When the F ratio was found to be significant, mean values were compared using a post hoc Bonferroni test. P<0.05 was considered to indicate significance in all analyses.
Results and Discussion
Phosphatidylserine externalization during membrane blebbing after exposure of macrophages to human neutrophil elastase (HNE) or substance P (SP) Phosphatidylserine (PS) is a major component of the inner layer of the plasma membrane, owing to membrane phospholipid asymmetry, and is externalized to the outer layer of the plasma membrane (Figure 1a) as membrane blebbing develops [23]. Neutrophil elastase, a PAR2 agonist, has been reported to activate microvesicle shedding from endothelial cells in a time-dependent manner [9]. In the present study, HNE significantly increased phosphatidylserine levels in cell-culture supernatant compared to untreated controls. Additionally, the PAR2 activating synthetic peptide, 2-Furoyl-LIGO-amide, induced a slight increase in PS. Surprisingly, a nonpeptide agonist (AC-264613) significantly upregulated PS levels rather than HNE (Figure 1b). We previously reported that AC-264613 significantly reduced p53 protein expression [24], and p53 activation is known to induce PS externalization early in apoptosis [25].
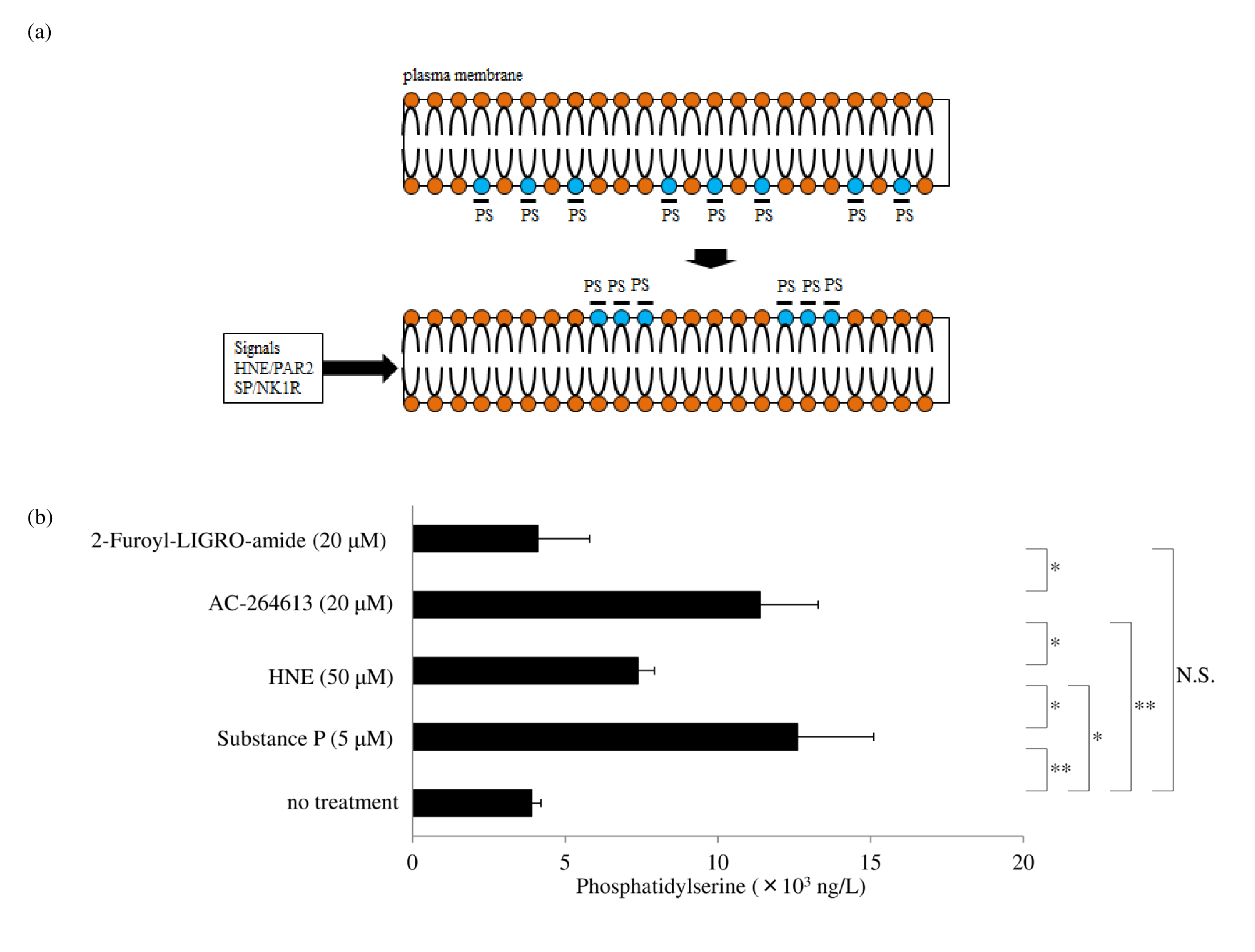
Studies reported that p53 deficiency in mouse embryonic fibroblasts also promotes Ras homolog family member A (RhoA)–Rho-associated protein kinase (ROCK)- dependent membrane blebs [26]. As for SP/neurokinin 1 receptor (NK1R) signaling, it has been reported that SP induces membrane blebbing in HEK293 cells [16]. We also previously reported that SP increased the release of membrane-bound tissue factor from human macrophages [27]. In addition, we reported that di(2- ethylhexyl) phthalate (DEHP) released tissue factorbearing microparticles from human macrophages and that these microparticles were phosphatidylserine positive in Western blotting. We also showed that phosphatidylserine-positivity of the culture supernatant was increased in a concentration-dependent manner after exposure of the macrophages to DEHP [28]. In this study, we found that exposure of macrophages to HNE or SP led to a significant increase in phosphatidylserine levels in cell-culture supernatants, as detected by ELISA. Most importantly, phosphatidylserine exposure has been reported to be required for shedding activation by a disintegrin and metalloprotease (ADAM)10/17 [11,12]. This led us to hypothesize that HNE or SP promotes the shedding activity of ADAM10/17 by phosphatidylserine externalization for the ectodomain shedding.
Activation of a disintegrin and metalloprotease (ADAM) by dual oxidase 2 (DUOX2)
The ADAM family induces proteolytic cleavage of transmembrane proteins. Members of the ADAM family are composed of a cytoplasmic domain and an extracellular metalloprotease domain with an attached pro-domain [29]. ADAM10/17 is composed of an extracellular metalloprotease (catalytic) domain and an intracellular cytoplasmic tail. Cysteine in the pro-domain is oxidized by reactive oxygen species (ROS) and reactive nitrogen species (RNS), and the pro-domain is consequently removed by cleavage from the catalytic domain, after which the metalloprotease domain is activated [30]. ROS includes hydrogen peroxide and superoxide, and RNS includes peroxynitrite and nitric oxide radical. Both hydrogen peroxide (H2O2) and peroxynitrite (ONOO-) activate ADAM10/17 metalloproteases. Mitochondrial ROS is a byproduct of oxidative metabolism, whereas nonmitochondrial ROS originates from extra-mitochondrial nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which is composed of membrane and cytosolic components. NADPH oxidase (NOX) is a ROS generating enzyme, and the NOX/dual oxidase (DUOX) family of NADPH oxides includes NOX1-5, DUOX1, and DUOX2. NADPH oxidase mediates production of a superoxide (O2-) ( Figure 2a), which produces superoxide dismutase (SOD) to generate hydrogen peroxide (H2O2). DUOX2 has both an NADPH oxidase domain and a peroxidase domain. Most importantly, the peroxidase domain produces peroxynitrite (ONOO−) through the interaction of nitrogen oxide with superoxide (Figure 2b). Therefore, DUOX2 activates ADAM10/17 by H2O2 and ONOO-.
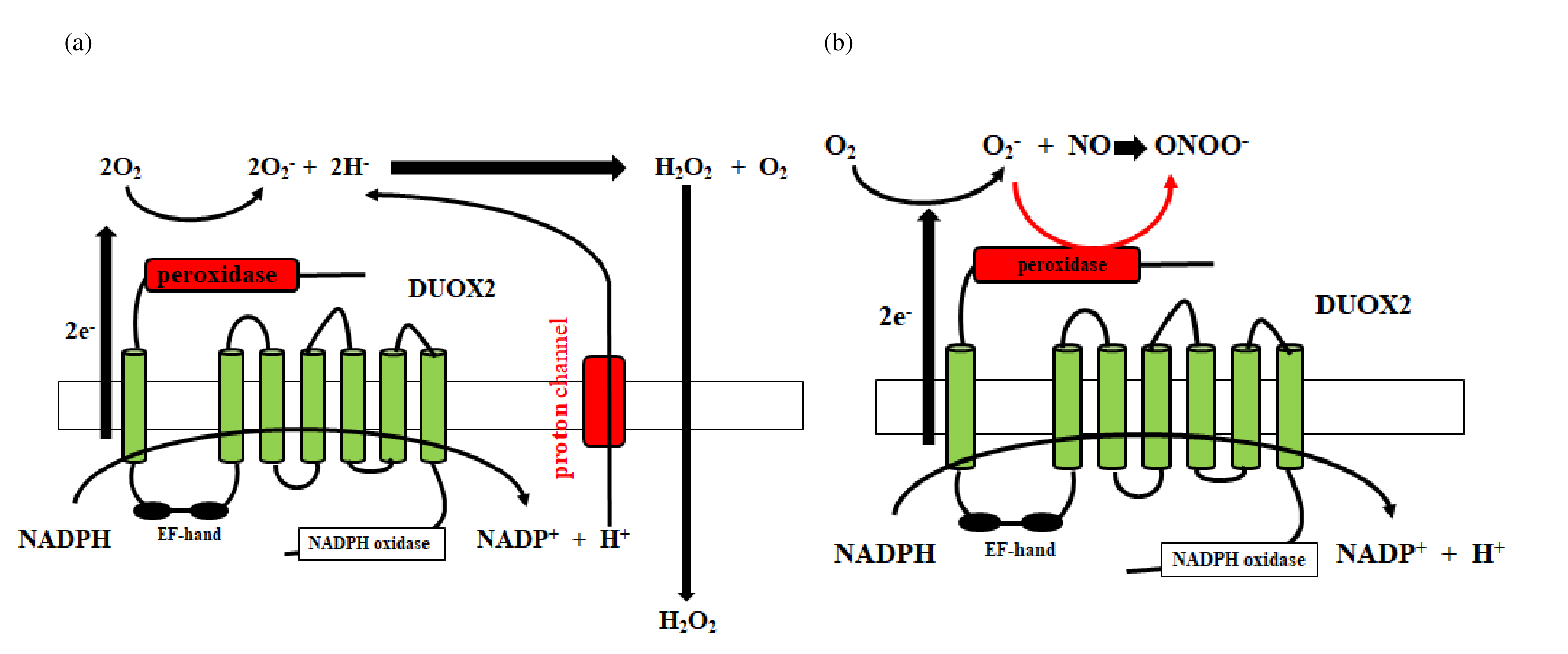
Differential peroxynitrite production in human macrophages by PAR2 or NK1R signaling
Nitric oxide synthase is activated by PAR2 signaling in endothelial cells [31]. Furthermore, neutrophil elastase inhibitor (Silvestat) has been reported to inhibit inducible nitric oxide synthase (iNOS) expression and NO production in hepatocytes [32]. The peroxidase-like domain of DUOX2 uses NADPH oxidase-mediated superoxide to generate the powerful oxidant peroxynitrite in response to NO [3].
Nitrotyrosine is known to be a marker for peroxynitrite generation [34]. Unexpectedly, we found that nitrotyrosine levels in the whole-cell lysate of human macrophages was not enhanced after exposure to HNE and PAR2 agonists (2-Furoyl-LIGO-amide and AC-264613) (Figure 3). SP/ NK 1R signaling promotes nitric oxide synthase 2/inducible nitric oxide synthase (NOS2/iNOS) mRNA expression [35] and enhances the production of NO in synovial fibroblasts of mice [36]. We found that protein levels of nitrotyrosine in whole-cell lysates were upregulated after exposure of human macrophages to SP, whereas those levels were significantly decreased in macrophages transfected with DUOX 2 siRNA after exposure to SP (Figure 3). This indicates that peroxynitirite production is differentially regulated in human macrophages by HNE/PAR2 and SP/ NK1R signaling.
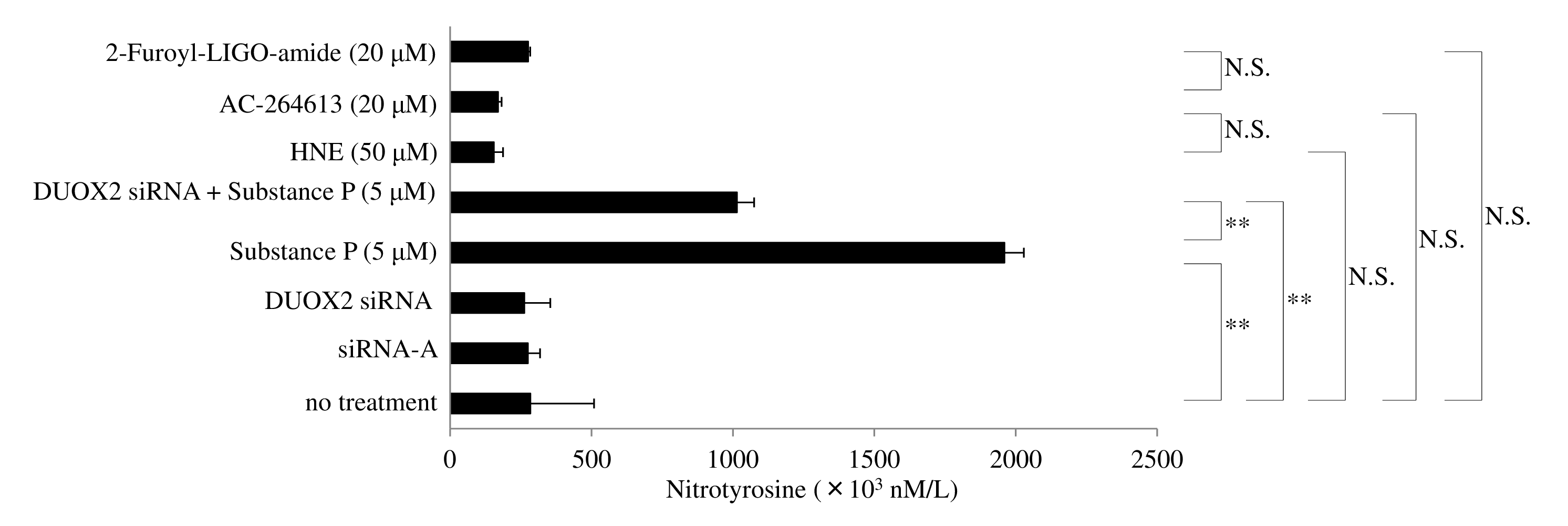
Transactivation of PAR2/EGFR/TLR4 by HNE/ PAR2 signaling
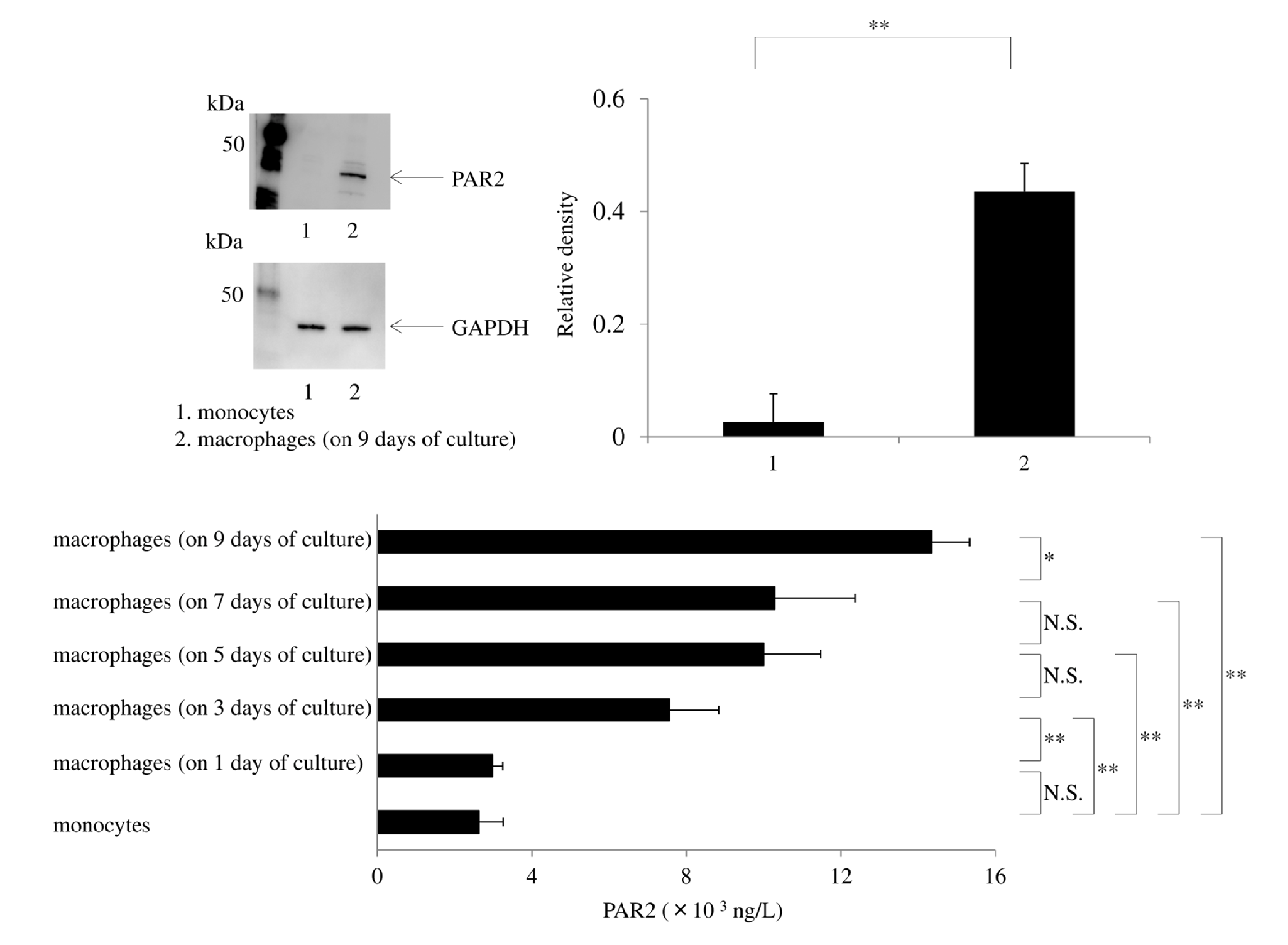
GM-CSF dependent macrophages (on day 9 of culture) significantly upregulated PAR2 expression compared with monocytes, when detected by western blotting (Figure 4a). Culture of macrophages with GM-CSF led to the upregulation of PAR2 production in a time-dependent manner. There was a significant increase in PAR2 from day 3 of the culture and PAR-2 protein levels were significantly higher on day 9 of culture than on day 7 (Figure 4b). Lipopolysaccharide enhanced IL-12p40 production by human macrophages. While HNE did not induce IL- 12p40 production, pretreatment of macrophages with HNE synergistically increased the IL-12p40 protein levels after LPS exposure. U73122 (a phospholipase C inhibitor) or Rottlerin (a PKC inhibitor) did not inhibit IL-12p40 production by macrophages exposed to only LPS, but either of these agents significantly attenuated IL-12p40 production by cells exposed to both HNE and LPS (Figures 5a-5c). Silencing of PAR2 with siRNA significantly reduced IL-12p40 production by macrophages stimulated with HNE and LPS. Silencing of β-arrestin 2, p22phox, or extracellular signal-regulated kinase 1/2 (ERK1/2) with siRNAs also attenuated IL-12p40 production by macrophages stimulated with HNE and LPS (Figures 5a and 5c). We previously reported that neutrophil elastase activates phospholipase C (PLC)/protein kinase C (PKC) signaling leads to cytokine production [37,38]. In addition, PLC/PKC signaling has been reported to modulate DUOX activity [39] (Figure 6a). The GPCR adaptor protein β-arrestin 2 modulates proinflammatory responses. We found that silencing β-arrestin 2 prevented an increase of IL-12p40 production by macrophages in response to stimulation with HNE plus LPS. Stimulation of PAR2 induces prolonged activation of ERK1/2 in a β-arrestin 2-dependent manner [40], and TRAF6 stimulates ERK activity [41].
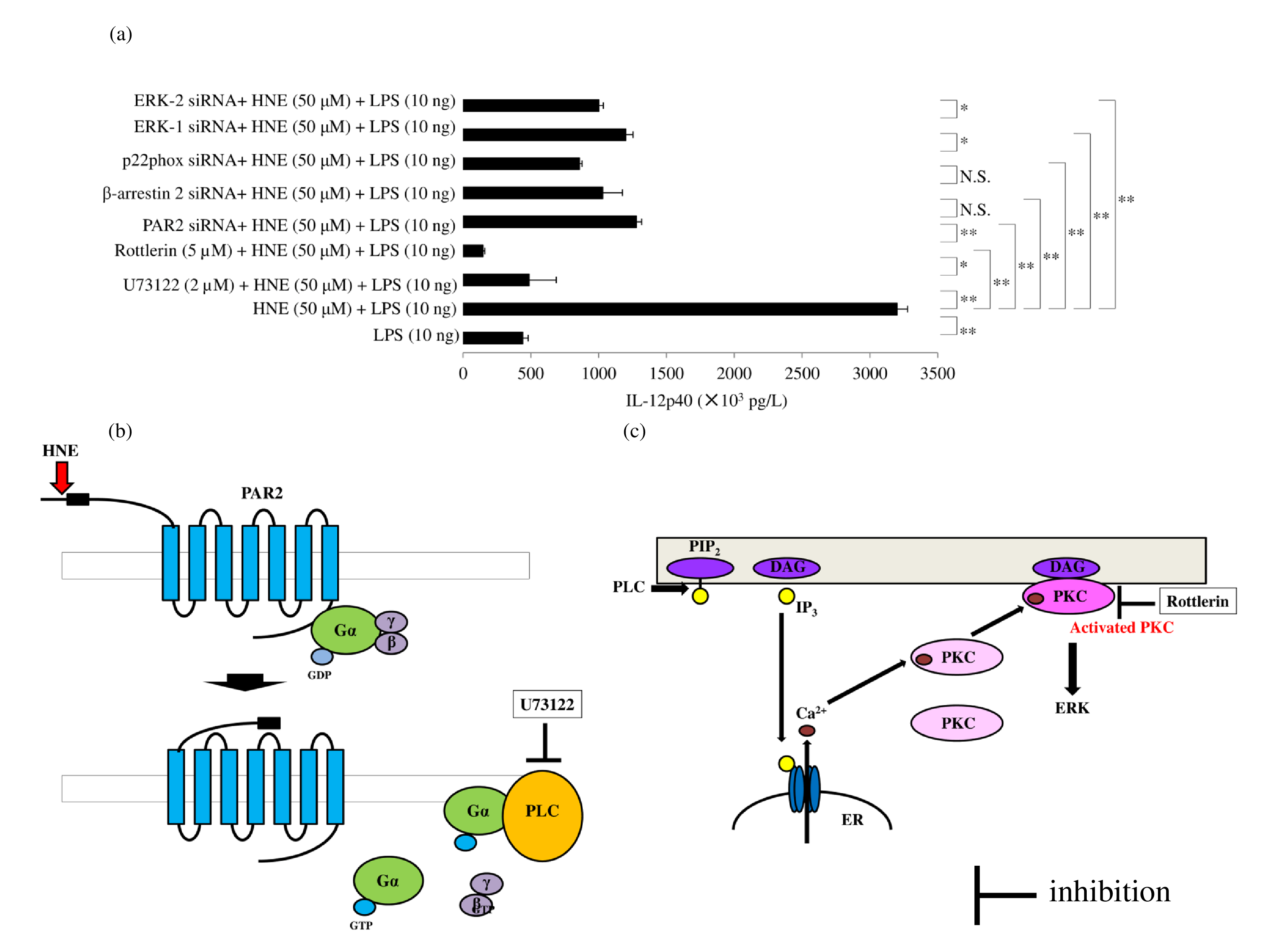
A schematic diagram represents the role of phospholipase C (PLC) (b) and protein kinase C (PKC) (c) on IL-12p40 production via the HNE/PAR2 signaling pathway. DUOX2 is activated by calcium and PKC (d). HNE: Human Neutrophil Elastase; PAR2: Protease-activated Receptor 2; GDP: Guanosine Diphosphate; Gα: G protein α-subunit; PLC: Phospholipase C; PIP2: Phosphatidylinositol 4,5-bisphosphate; DAG: Diacyl Glycerol, IP3: Inositol 1,4,5-trisphosphate, ER: Endoplasmic Reticulum, Ca: Calcium, PKC: Protein Kinase C; ERK: Extracellular Signal-regulated Kinase.
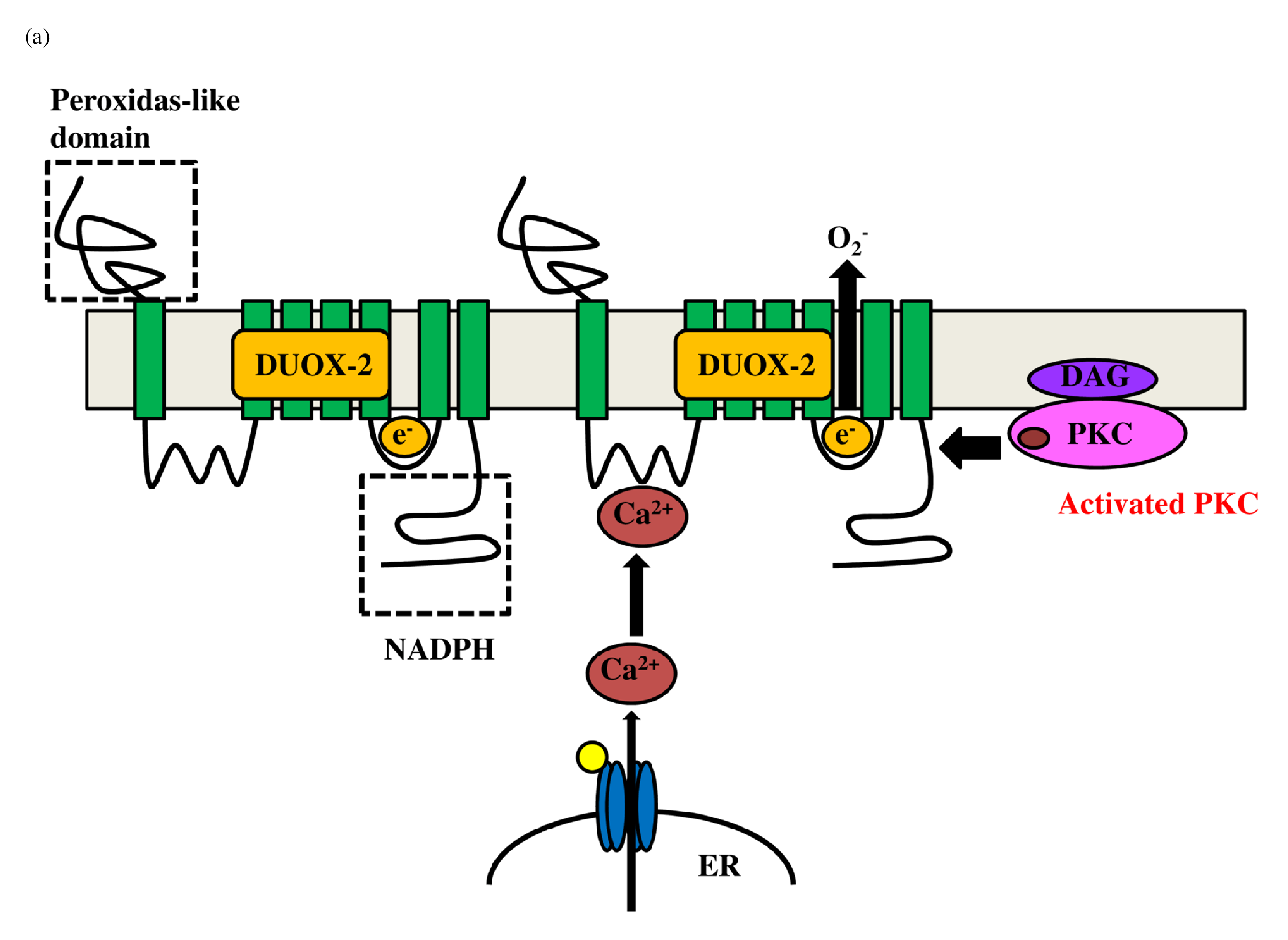
The schematic diagram represents activation of DUOX2 by PKC and Ca2+ (a). NADPH oxidase composed of p22phox, Gp91phox, p67phox and p40phox produces hydrogen peroxide (H2O2) (c). Transactivation of DUOX2/ADAMs/EGFR is induced by DUOX2-derived H2O2 (d). Finally, EGFR kinase activates TLR4 (e). DUOX: dual oxidase, NADPH: nicotinamide adenine dinucleotide phosphate, NADP: Nicotinamide Adenine Dinucleotide Phosphate; EGFR: Epidermal Growth Factor Receptor; ADAM: A Disintegrin and Metalloprotease, LPS: Lipopolysaccharide, TLR: Toll-like Receptor; CD14: Cluster of Differentiation 14, MD2: Myeloid Differentiation Factor 2; MyD88: Myeloid Differentiation Factor 88.
Next, we investigated the role of ERK in the enhancement of IL-12p40 production by HNE plus LPS. Silencing of ERK1/2 also significantly reduced IL-12p40 production by stimulated macrophages. ERK mediates important downstream aspects of EGFR signaling, and phosphorylation of this receptor at threonine-669 by ERK influences both signaling and trafficking [42]. The β-arrestin 2 scaffold protein mediates the proinflammatory effects of PAR2 and promotes ERK1/2 phosphorylation [43]. Silencing p22phox attenuated the enhancement of IL-12p40 production by macrophages stimulated with HNE plus LPS. Activation of PAR2 leads to upregulation of the DUOX2/ROS pathway [44]. We found that silencing DUOX 2 with siRNA significantly reduced the production of IL-12p40 by macrophages stimulated with HNE plus LPS (Figure 6b). Reactive oxygen species produced through NADPH oxidase mediate EGFR activation, and p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system [45] (Figure 6c). It has also been reported that silencing p22phox prevents EGFR activation [46]. Indeed, we found that silencing p22phox blunted the enhancement of IL-12p40 production by macrophages in response to HNE plus LPS. Therefore, PAR 2-mediated EGFR transactivation is dependent on H 2O2 derived from DUOX2 activation. Transfection of macrophages with small interfering RNA for DUOX2, EGFR, or TLR4 significantly blunted the increase of IL- 12p40 in response to treatment with HNE plus LPS. PAR2 mediates upregulation of DUOX2 [44]. ADAM17- mediated shedding of EGFR ligands induces EGFR activation [47] (Figure 6d). The pro-domain attached to the metalloprotease domain regulates the activity of ADAM 10/17 sheddase [48]. Indeed, a recombinant prodomain was reported to inhibit the activation of ADAM17 [ 49]. Activation of ADAM10/17 requires detachment of the pro-domain from the metalloprotease domain [29]. This detachment is triggered by oxidation of cysteine in the pro-domain in response to reactive oxygen species (ROS) and reactive nitrogen species (RNS) [30]. The oxidation of cysteine in the pro-domain is mediated by H2O2 [50] or peroxynitrite [51]. After EGFR activation, EGFR kinase activity is necessary for TLR4 signaling [52] (Figure 6e). We found that small interfering RNA for p22phox, an NADPH oxidase subunit, reduced IL-12p40 levels in response to HNE and LPS. Therefore, PAR2/EGFR/TLR4 transactivation is regulated by H2O2 derived from DUOX2 activation in IL-12p40 production by human macrophages in response to HNE and LPS.
Ectodomain shedding of fractalkine by DUOX2 signaling
Innate immunity may play an important role in the initiation and/or progression of autoimmune diseases. Chemokines are critical for the innate immune response and are involved in the selective recruitment of immune cells and their migration to the inflamed tissues. The chemokine fractalkine, also known as CX3CL1, is induced in a membrane-bound form in macrophages, fibroblasts, endothelial, and dendritic cells [53]; however, serum levels of soluble fractalkine are known to be elevated in autoimmune diseases [18]. Studies have indicated that fractalkine shedding is mediated by the sheddase ADAM [ 19]. We focused on the regulatory mechanism of shedding for membrane-bound fractalkine. Treatment of human macrophages with SP upregulated levels of membranebound fractalkine. Interestingly, small interfering RNA for DUOX 2 further increased membrane-bound fractalkine, as determined by ELISA (Figure 7a) and western blotting ( Figure 7c), but decreased soluble fractalkine compared with cells treated with SP alone (Figure 7b). This suggests that DUOX2/ADAM10/17 activation mediates shedding of membrane-bound fractalkine.
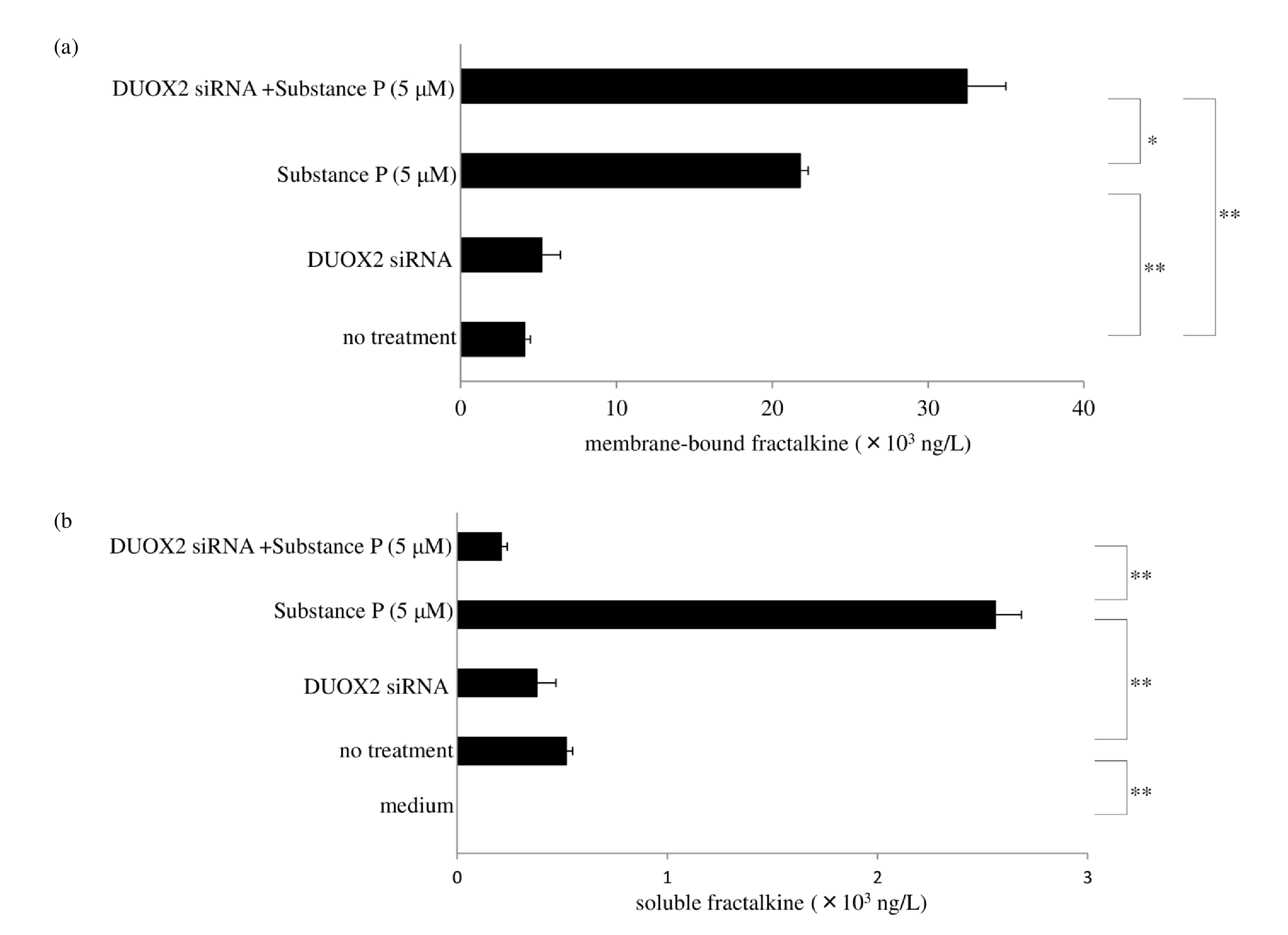
Protein levels of membrane-bound fractalkine were determined by western blotting. Data were obtained by using samples from three volunteers in each group, and a representative result is shown as arbitrary density units (c).
Regulation of ectodomain shedding by the ubiquitin-proteasome system
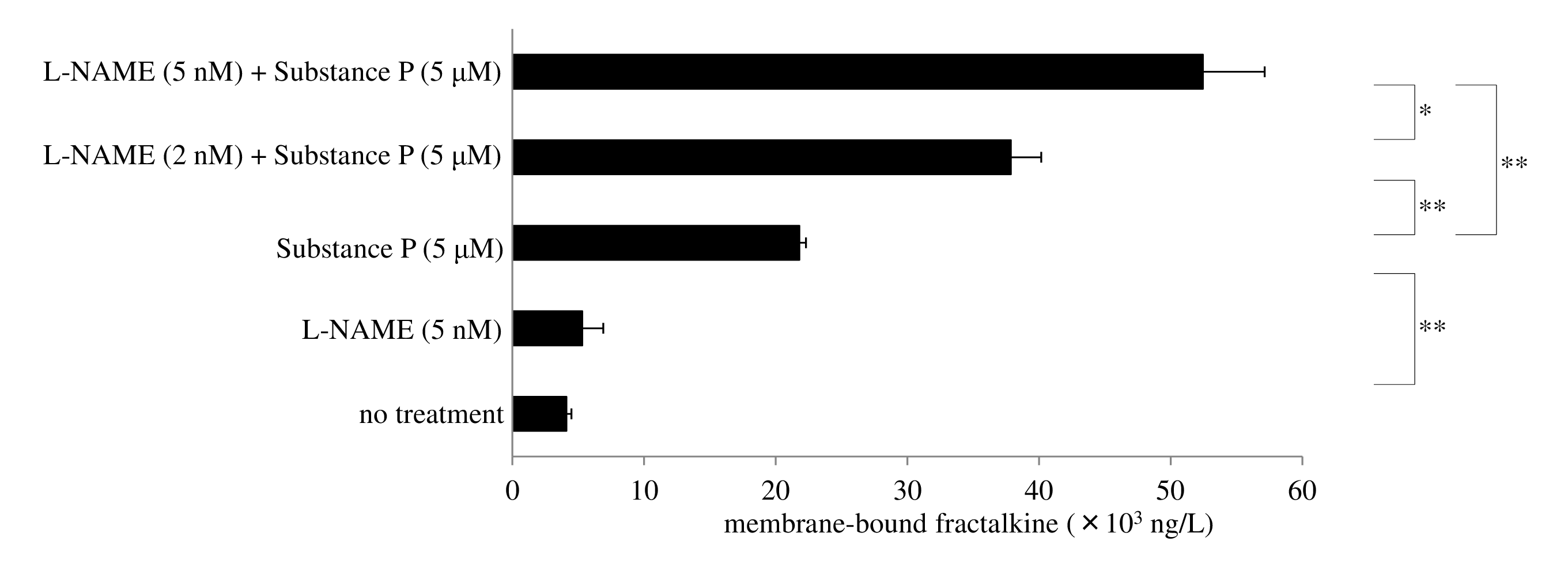
The ubiquitin-proteasome system catabolizes proteins in the cytosol and nucleus, and plays a pivotal role in inflammatory and autoimmune diseases [54]. Ectodomain shedding of fractalkine is mediated by DUOX2 activation and increases soluble fractalkine rather than membranebound form. We found that N(ω)-nitro-L-arginine methyl ester (L-NAME, a nitric oxide synthase inhibitor) decreased protein levels of nitrotyrosine and concomitantly increased expression of membrane-bound fractalkine after exposure of human macrophages to SP (Figure 8). Shedding of fractalkine is promoted by ADAM10/17 activation through DUOX2-mediated peroxinitrite generation in response to NO.
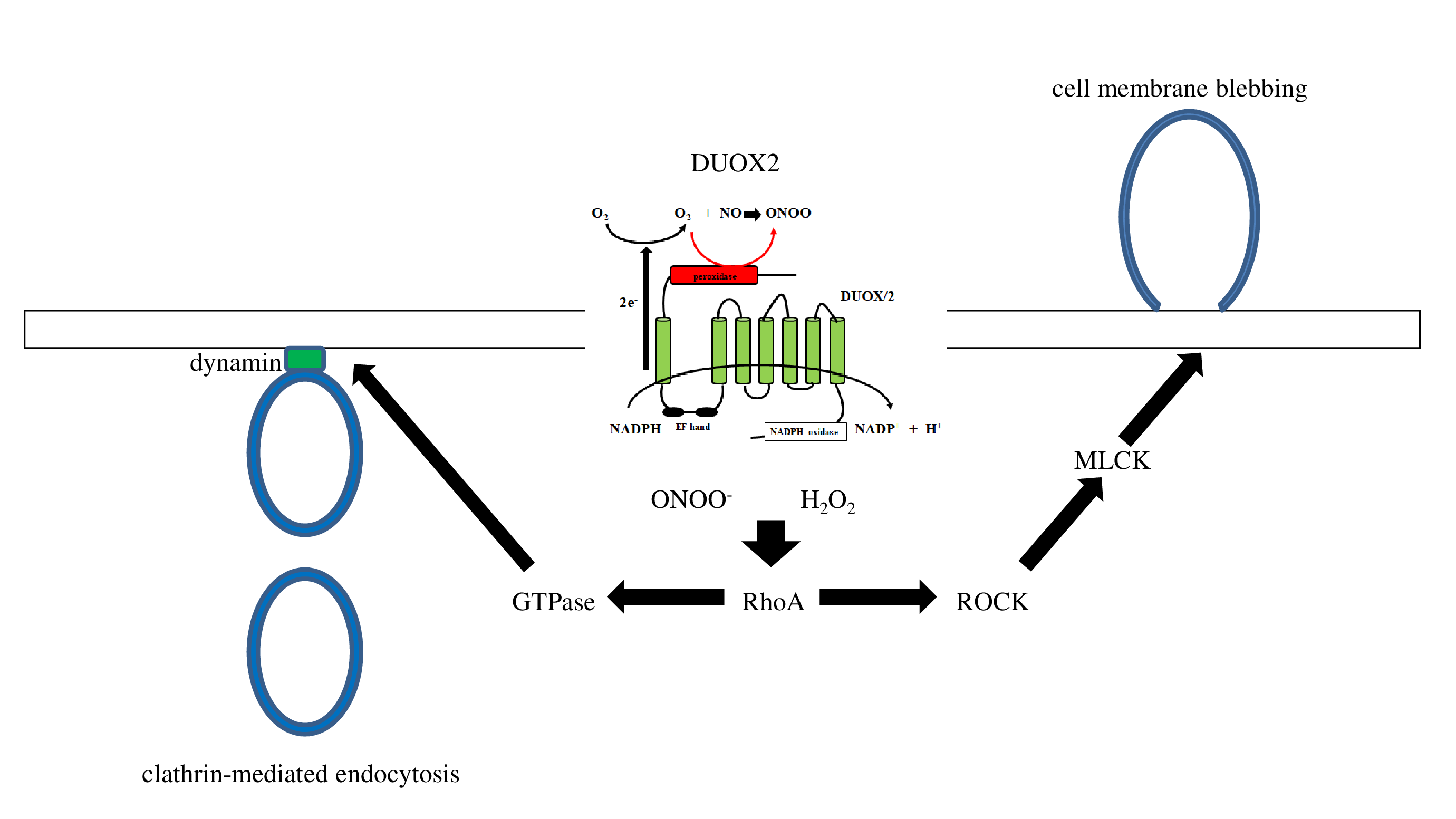
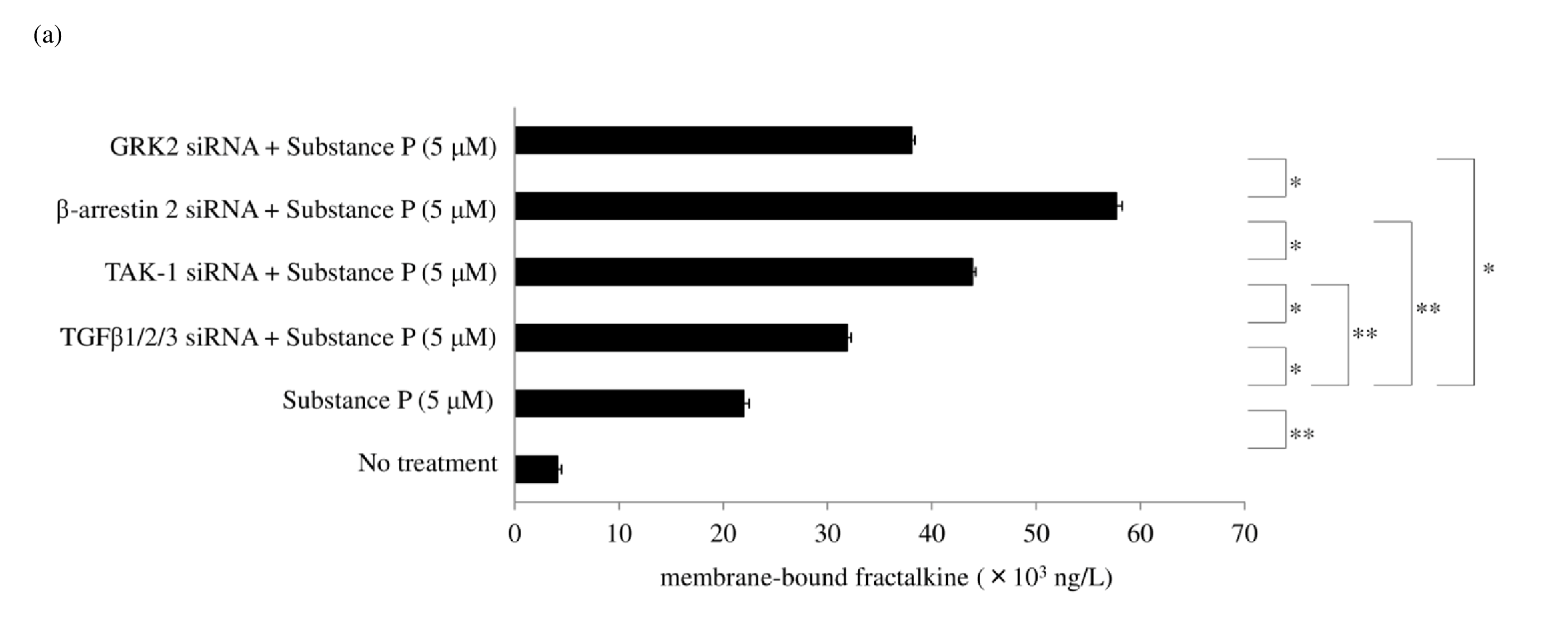
SP/NK1R signaling activates Gαq, leading to Ca2+ mobilization and activation of protein kinase C, and protein kinase C upregulates NOS2/iNOS induction of NO synthesis [36,55]. Gaq signaling recruits β arrestin 2 to NK1R, and β arrestin 2 activates PI3K-Akt [56], leading to the induction of NOS2/iNOS expression [57]. Thus, β arrestin 2 has been reported to lead to NOS2/iNOS induction [58]. NK1R recycling and resensitization require its internalization after SP stimulation. Understanding the underlying molecular mechanisms of SP-induced internalization of NK 1R is important because of the essential role of signal transduction during NK1R trafficking. NK1R signaling is derived not only from the plasma membrane but also from the endosomal membrane during internalization. DUOX2 activation may be associated with multiple cellular events, such as membrane blebbing and GPCR endocytosis. Rhoassociated coiled-coil–containing protein kinase (ROCK) activity is required for membrane blebbing [59]. NK1R activation induces membrane blebbing via the Rho/ROCK/ MLCK pathway [27,60]. Peroxynitrite and hydrogen peroxide also enhance RhoA, ROCK1, and ROCK2 activity [ 61,62]. GPCRs are dephosphorylated through clathrinmediated endocytosis and recycled back to the cell surface. The process of membrane fission of vesicles is dependent on dynamin, a membrane-remodeling GTPase. The Rho family of GTPases also is a regulator for multiple cellular events. These observations suggest that RhoA activated by DUOX2-derived ROS and RNS is associated with the membrane fission of endosomes. Figure 9 shows a possible role of DUOX2 for cell membrane blebbing and clathrin-mediated endocytosis. β-arrestin 2 also serves as an adaptor during SP-mediated internalization of NK1R [ 63]. We found that siRNA for β arrestin 2 significantly increased membrane-bound fractalkine after exposure to SP. G-protein-coupled receptor kinase 2 (GRK2) phosphorylates NK1R and recruits β arrestin 2. RNA interference of GRK2 also increased membrane-bound fractalkine levels as measured by ELISA (Figure 10a) and western blotting (Figure 10b). NOS2/iNOS mRNA and protein expression in macrophages was also reported to be inhibited by TGFβ1 [55]. This led us to hypothesize that TGFβ1 may regulate peroxynitrite generation and shedding of fractalkine. Our results showed that TGFβ1/2/3 siRNA led to a slight increase in membrane-bound fractalkine. As for the signaling pathway of TGFβ1, silencing of RNA for TAK-1 upregulated membrane-bound fractalkine (Figure 10a), but silencing of RNA for the Smad family did not. TAK-1 is known to induce NOS2/iNOS expression [56], but Smad signaling has been reported to negatively regulate NOS2/iNOS induction [57]. This led us to hypothesize that enhanced levels of TGFβ1 attenuate NO production and lead to a decrease in peroxynitrite generation by DUOX 2. We found that RNA interference of transcription factor specificity protein 1 (Sp1) increased in TGFβ1 but was not sufficient to reduce NOS2/iNOS production or to inhibit the shedding of membrane-bound fractalkine. The transcription factor CCAAT-enhancer binding protein (C/ EBPβ) was previously reported to mediate NOS2/iNOS production [64]; however, we found that C/EBPβ siRNA did not affect NOS2/iNOS expression after exposure to SP.
Protein levels of membrane-bound fractalkine were determined by western blotting. Data were obtained by using samples from three volunteers in each group, and a representative result is shown as arbitrary density units (b).
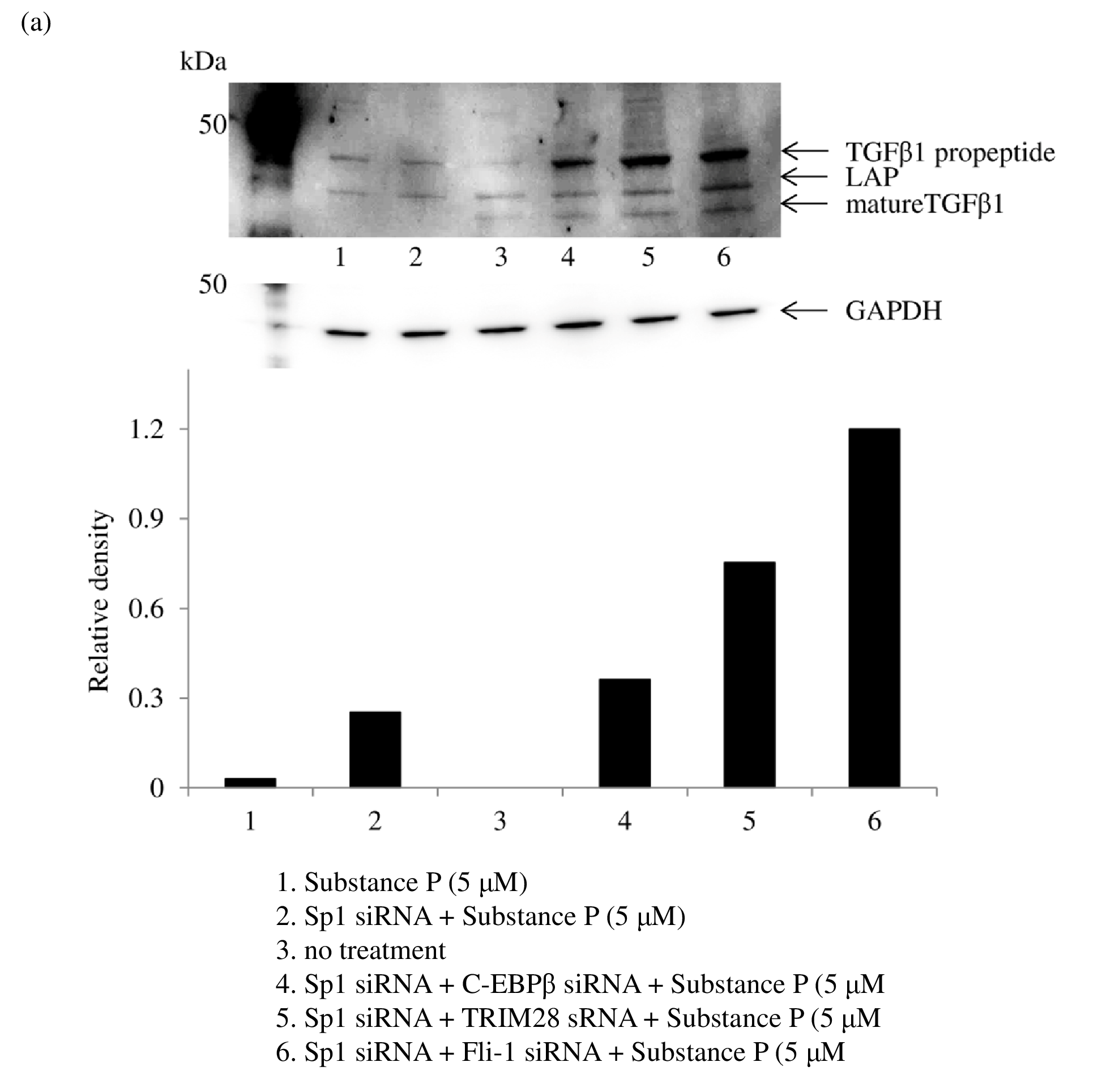
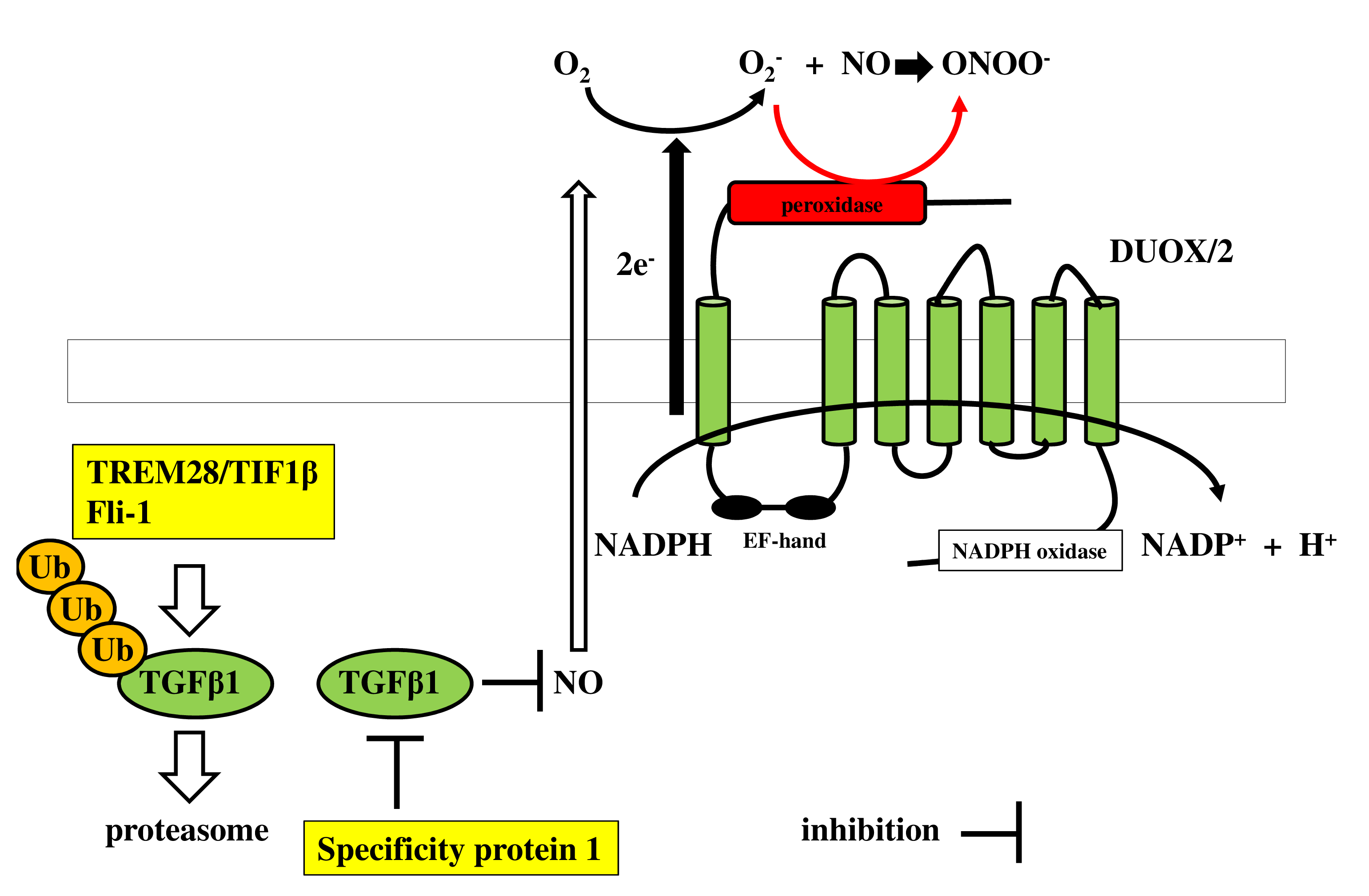
The ubiquitin-proteasome system plays a pivotal role in inflammatory and autoimmune diseases [54]. Ubiquitination is mediated by E3 ubiquitin ligase, which also regulates TGFβ1 levels by ubiquitination and proteasome degradation [65]. Tripartite motif 28 (TRIM28)/transcriptional intermediary factor 1β (TIF1β) functions as an E3 ubiquitin ligase [66]. The Friend leukemia integration 1 (Fli-1) transcription factor is reported to be associated with autoimmune diseases such as systemic lupus erythematosus and systemic sclerosis [67]. Fli-1 regulates murine double minute 2 (MDM2) expression [68], which also acts as an E3 ubiquitin ligase [ 69]. Hence, this led us to hypothesize that TRIM28/ TIF1β or Fli-1 modulates ADAM10/17 sheddase activity by ubiquitin-proteasomal degradation of TGFβ1. Therefore, we attempted to modulate ubiquitin-proteasome degradation of TGFβ1 by ubiquitin ligase. Surprisingly, double transfection of Sp1 siRNA and TRIM28/TIF1β siRNA or Fli-1 siRNA led to a further significant increase in mRNA (Figure 11a) and protein levels of TGFβ1 (Figure 11b), resulting in a consistent reduction of NOS2/iNOS levels and upregulation of membrane-bound fractalkine expression after exposure to SP (Figures 11c and 11d). Figure 12 depicts a regulatory mechanism of peroxynitrite production by ubiquitin-proteasome system.
Conclusion
Membrane-embedded DUOX2 has NADPH and peroxidase domains. NADPH oxidase induces hydrogen peroxide (H2O2), while the peroxidase domain produces peroxynitrite (ONOO−) through the interaction of NO with superoxide. Both H2O2 and ONOO− activate ADAM10/17 metalloproteases. HNE/PAR2 signaling transactivates DUOX2/EGFR/TLR4 in IL-12p40 production, which is H2O2 dependent for ADAMs activation. SP/NK1R signaling induces ONOO− production, which activates ADAMs for fractalkine shedding. Ectodomain shedding by GPCRs occurs either by H2O2 or H2O2 plus ONOO− via DUOX2 activation.
Consent for Publication
I understand that the text and any pictures published in the article will be freely available on the internet and may be seen by the general public.
Conflict of Interest
The authors declare that they have no conflict of interest.
Acknowledgement
This study was partly supported by a Kumamoto Health Science University special fellowship grant (No. 2018-C- 2).
Author Contribution Statement
Rui Yamaguchi: Conceived and designed the experiments, wrote the paper. Arisa Sakamoto: Performed the experiments. Reona Yamaguchi: Analysis tools or data. Misa Haraguchi: Performed the experiments. Shinji Narahara: Performed the experiments. Hiroyuki Sugiuchi: Materials, analysis tools or data. Yasuo Yamaguchi: Wrote the paper.
References
2. Aoki M, Yamaguchi R, Yamamoto T, Ishimaru Y, Ono T, Sakamoto A, et al. Granulocyte-macrophage colonystimulating factor primes interleukin-13 production by macrophages via protease-activated receptor-2. Blood Cells Mol Dis. 2015;54:353-359.
3. Boots AW, Gerloff K, Bartholomé R, van Berlo D, Ledermann K, Haenen GR, et al. Neutrophils augment LPS-mediated pro-inflammatory signaling in human lung epithelial cells. Biochim Biophys Acta. 2012;1823:1151- 1162.
4. Matsuse H,Yanagihara K, Mukae H, Tanaka K, Nakazato M, Kohno S. Association of plasma neutrophil elastase levels with other inflammatory mediators and clinical features in adult patients with moderate and severe pneumonia. Respir Med. 2007;101:1521-1528.
5. Hirche TO, Benabid R, Deslee G, Gangloff S, Achilefu S, Guenounou M, et al. Neutrophil elastase mediates innate host protection against Pseudomonas aeruginosa. J Immunol. 2008;181:4945-4954.
6. Benabid R, Wartelle J, Malleret L, Guyot N, Gangloff S, Lebargy F, et al. Neutrophil elastase modulates cytokine expression: contribution to host defense against Pseudomonas aeruginosa-induced pneumonia. J Biol Chem. 2012;287:34883-34894.
7. Belaaouaj A, McCarthy R, Baumann M, Gao Z, Ley TJ, Abraham, S.N, et al. Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat Med. 1998;4:615-618.
8. Rallabhandi P, Nhu QM, Toshchakov VY, Piao W, Medvedev A.E,Hollenberg MD, et al. Analysis of proteinaseactivated receptor 2 and TLR4 signal transduction: a novel paradigm for receptor cooperativity. J BiolChem. 2008;283:24314-24325.
9. Alfaidi M, Wilson H, Daigneault M, Burnett A, Ridger V, Chamberlain J, et al. Neutrophil Elastase Promotes interleukin-1β Secretion From Human Coronary Endothelium. J Biol Chem. 2015;290:24067-24078.
10. Bratton DL, Fadok VA, Richter DA, Kailey JM, Guthrie LA, Henson PM. Appearance of phosphatidylserine on apoptotic cells requires calcium-mediated nonspecific flipflop and is enhanced by loss of the aminophospholipid translocase. J Biol Chem. 1997;272:26159-26165.
11. Sommer A, Kordowski F, Büch J, Maretzky T, Evers A, Andrä J, et al. Phosphatidylserine exposure is required for ADAM17 sheddase function. Nat Commun. 2016;7:11523.
12. Reiss K, Bhakdi S. The plasma membrane: Penultimate regulator of ADAM sheddase function. Biochim Biophys Acta Mol Cell Res. 2017;1864:2082-2087.
13. Sanchez-Guerrero E, Chen E, Kockx M, An SW, Chong BH, Khachigian LM. IL-1beta signals through the EGF receptor and activates Egr-1 through MMP-ADAM. PLoS One. 2012;7:e39811.
14. Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, et al. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884-888.
15. Yamaguchi R, Yamamoto T, Sakamoto A, Narahara S, Sugiuchi H, Yamaguchi Y. Neutrophil elastase enhances IL-12p40 production by lipopolysaccharide-stimulated macrophages via transactivation of the PAR-2/EGFR/ TLR4 signaling pathway. Blood Cells Mol Dis. 2016;59:1-7.
16. Meshki J, Douglas SD, Lai JP, Schwartz L, Kilpatrick LE, Tuluc F. Neurokinin 1 receptor mediates membrane blebbing in HEK293 cells through a Rho/Rho-associated coiled-coil kinase-dependent mechanism. J Biol Chem. 2009;284:9280-9289.
17. Barbosa-Cobos RE, Lugo-Zamudio G, Flores-Estrada J, Becerril-Mendoza LT, Rodríguez-Henríquez P, Torres- González R, et al. Serum substance P: an indicator of disease activity and subclinical inflammation in rheumatoid arthritis. Clin Rheumatol. 2018;37: 901-908.
18. Yajima N, Kasama T, Isozaki T, Odai T, Matsunawa M, Negishi M, et al. Elevated levels of soluble fractalkine in active systemic lupus erythematosus: potential involvement in neuropsychiatric manifestations. Arthritis Rheum. 2005;52:1670-1675.
19. Garton KJ, Gough PJ, Blobel CP, Murphy G, Greaves DR, Dempsey PJ, et al. Tumor necrosis factor-alphaconverting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1). J Biol Chem. 2001;276:37993-38001.
20. Yamaguchi R, Haraguchi M, Yamaguchi R, Sakamoto A, Narahara S, Sugiuchi H, Yamaguchi Y. TRIM28/TIF1β and Fli-1 negatively regulate peroxynitrite generation via DUOX2 to decrease the shedding of membrane-bound fractalkine in human macrophages after exposure to substance P. Cytokine. 2020;134:155180.
21. Strieter RM, Remick DG, Lynch J.P.3rd, Genord M, Raiford C, Spengler R, et al. Differential regulation of tumor necrosis factor-alpha in human alveolar macrophages and peripheral blood monocytes: a cellular and molecular analysis. Am. J. Respir. Cell. Mol. Biol. 1989;1:57-63.
22. Yamaguchi R, Yamamoto T, Sakamoto A, Ishimaru Y, Narahara S, Hirose E, et al. Mechanism of interleukin-13 production by granulocyte-macrophage colonystimulating factor-dependent macrophages via proteaseactivated receptor-2. Blood Cells Mol. Dis. 2015;55:21-26.
23. Kirov A, Al-Hashimi H, Solomon P, Mazur C, Thorpe PE, Sims PJ, et al. Phosphatidylserine externalization and membrane blebbing are involved in the nonclassical export of FGF1. J Cell Biochem. 2012:113:956-966.
24. Yamaguchi R, Yamamoto T, Sakamoto A, IshimaruY, Narahara S, Sugiuchi H, et al. A protease-activated receptor 2 agonist (AC-264613) suppresses interferon regulatory factor 5 and decreases interleukin-12p40 production by lipopolysaccharide-stimulated macrophages: Role of p53. Cell Biol Int. 2016;40:629-641.
25. Geske FJ, Lieberman R, Strange R, Gerschenson LE.. Early stages of p53-induced apoptosis are reversible. Cell Death Differ. 2001;8:182-191.
26. Gadea G, de Toledo M, Anguille C, Roux P. Loss of p53 promotes RhoA–ROCK-dependent cell migration and invasion in 3D matrices. J Cell Biol. 2007;178:23-30.
27. Yamaguchi R, Yamamoto T, Sakamoto A, Ishimaru Y, Narahara S, Sugiuchi H, et al. Substance P enhances tissue factor release from granulocyte-macrophage colony-stimulating factor-dependent macrophages via the p22phox/β-arrestin 2/Rho A signaling pathway. Blood Cells Mol Dis. 2016;57:85-90.
28. Yamaguchi R, Sakamoto A, Yamaguchi R, Haraguchi M, Narahara S, Sugiuchi H, et al. Di-(2-Ethylhexyl) Phthalate Promotes Release of Tissue Factor-Bearing Microparticles From Macrophages via the TGFβ1/Smad/ PAI-1 Signaling Pathway. Am J Med Sci. 2019;357:492- 506.
29. Reiss K, Saftig P. The “a disintegrin and metalloprotease” (ADAM) family of sheddases: physiological and cellular functions. Semin Cell Dev Biol.2009;20:126-137.
30. Sanderson MP, Dempsey PJ, Dunbar AJ. Control of ErbB signaling through metalloprotease mediated ectodomain shedding of EGF-like factors. Growth Factors. 2006;24:121-136.
31. Maruyama K, Kagota S, McGuire JJ, Wakuda H, Yoshikawa N, Nakamura K, et al. Enhanced Nitric Oxide Synthase Activation via Protease-Activated Receptor 2 Is Involved in the Preserved Vasodilation in Aortas From Metabolic Syndrome Rats. J Vasc Res. 2015;52:232-243.
32. Araki Y, Matsumiya M, Matsuura T, Kaibori M, Okumura T, Nishizawa M, et al. Sivelestat Suppresses iNOS Gene Expression in Proinflammatory Cytokine- Stimulated Hepatocytes. Dig Dis Sci. 2011;56:1672-1681.
33. Robinson KM, Beckman JS. Synthesis of peroxynitrite from nitrite and hydrogen peroxide. Methods Enzymol. 2005;396:207-214.
34. Kuhn DM, Sakowski SA, Sadidi M, Geddes TJ. Nitrotyrosine as a Marker for Peroxynitrite-Induced Neurotoxicity: The Beginning or the End of the End of Dopamine Neurons? J Neurochem. 2004;89:529-536.
35. Jeon HK, Jung NP, Choi IH, Oh YK, Shin HC, Gwag BJ. Substance P augments nitric oxide production and gene expression in murine macrophages. Immunopharmacology. 1999;41:219-226.
36. O’Shaughnessy MC, Vetsika EK, Inglis JJ, Carleson J, Haigh R, Kidd BL, et al. The effect of substance P on nitric oxide release in a rheumatoid arthritis model. Inflamm Res. 2006;55:236-240.
37. Ishihara K, Yamaguchi Y, Uchino S, Furuhashi T, Yamada S, Kihara S, et al. ICAM-1 signal transduction in cells stimulated with neutrophil Elastase. Dig Dis Sci. 2006;51:2102-2112.
38. Kawata J, Aoki M, Ishimaru Y, Ono T, Sagara K, Narahara S, et al. Mechanism of tissue factor production by monocytes stimulated with neutrophil elastase. Blood Cells Mol Dis. 2015;54:206-209.
39. Song Y, Ruf J, Lothaire P, Dequanter D, Andry G, Willemse E, et al. Association of duoxes with thyroid peroxidase and its regulation in thyrocytes. J Clin Endocrinol Metab. 2010;95:375-382.
40. Tohgo A, Pierce KL, Choy EW, Lefkowitz RJ, Luttrell LM. beta-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J Biol Chem. 2002;277:9429-9436.
41. Kashiwada M, Shirakata Y, Inoue JI, Nakano H, Okazaki K, Okumura K, et al. Tumor necrosis factor receptor-associated factor 6 (TRAF6) stimulates extracellular signal-regulated kinase (ERK) activity in CD40 signaling along a ras-independent pathway. J Exp Med. 1998;187:237-244.
42. Li X. Huang Y, Jiang J, Frank SJ. ERK-dependent threonine phosphorylation of EGF receptor modulates receptor downregulation and signaling. Cell Signal. 2008;20:2145-2155.
43. Nichols HL, Saffeddine M, Theriot BS, Hegde A, Polley D, El-Mays T, et al. β-Arrestin-2 mediates the proinflammatory effects of proteinase-activated receptor-2 in the airway. Proc Natl Acad Sci U S A. 2012;109:16660- 16665.
44. Nadeem A, Alharbi NO, Vliagoftis H, Tyagi M, Ahmad SF, Sayed-Ahmed MM. Proteinase activated receptor- 2-mediated dual oxidase-2 up-regulation is involved in enhanced airway reactivity and inflammation in a mouse model of allergic asthma. Immunology. 2015;145:391-403.
45. Ushio-Fukai M, Zafari AM, Fukui T, Ishizaka N, Griendling KK.p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells. J Biol Chem. 1996;271:23317-23321.
46. Cattaneo F, Iaccio A, Guerra G, Montagnani S, Ammendola R. NADPH-oxidase-dependent reactive oxygen species mediate EGFR transactivation by FPRL1 in WKYMVm-stimulated human lung cancer cells. Free Radic Biol Med. 2011;51:1126-1136.
47. Blobel CP. ADAMs: Key Components in EGFR Signalling and Development. Nat Rev Mol Cell Biol. 2005;6:32-43.
48. Moss ML, Bomar M, Liu Q, Sage H, Dempsey P, Lenhart PM, et al. The ADAM10 prodomain is a specific inhibitor of ADAM10 proteolytic activity and inhibits cellular shedding events. J Biol Chem. 2007;282:35712- 35721.
49. Li X, Yan Y, Huang W, Yang Y. The study of the inhibition of the recombinant TACE prodomain to endotoxemia in mice. Int J Mol Sci. 2009;10:5442-5454.
50. Zhang Z, Oliver P, Lancaster JRJr, Schwarzenberger PO, Joshi MS, Cork J, et al. Reactive oxygen species mediate tumor necrosis factor alpha-converting, enzymedependent ectodomain shedding induced by phorbol myristate acetate. FASEB J. 2001;15:303-305.
51. Zeida A, González Lebrero MC, Radi R, Trujillo M, Estrin DA. Mechanism of cysteine oxidation by peroxynitrite: An integrated experimental and theoretical study. Arch Biochem Biophys. 2013;539:81-86.
52. Chattopadhyay S, Veleeparambil M, Poddar D, Abdulkhalek S, Bandyopadhyay SK, Fensterl V, et al. EGFR Kinase Activity Is Required for TLR4 Signaling and the Septic Shock Response. EMBO Rep. 2015;16:1535-1547.
53. Ruth JH, Volin MV, Haines GK3rd, Woodruff DC, Katschke KJJr, Woods JM, et al. Fractalkine, a novel chemokine in rheumatoid arthritis and in rat adjuvantinduced arthritis. Arthritis Rheum. 2001;44:1568-1581.
54. Wang TY, Li J, Jin Z, Wu F, Zhou Q. Inhibitory effect of TGF-β1 on NO production in peritoneal macrophages from collagen-induced arthritis rats involving the LPSTLR4 pathway. Mol Med Rep. 2013;8:1143-1148.
55. Su K, Zeng P, Liang W, Luo Z, Wang Y, Lv X, et al. FTY720 Attenuates Angiotensin II- Induced Podocyte Damage via Inhibiting Inflammatory Cytokines. Mediators of inflammation. 2017 Feb 7;2017:3701385.
56. Bhat NR, Shen Q, Fan F. TAK1-mediated induction of nitric oxide synthase gene expression in glial cells. J Neurochem. 2003;87:238-247.
57. Sugiyama Y, Kakoi K, Kimura A, Takada I, Kashiwagi I, Wakabayashi Y, et al. A. Smad2 and Smad3 are redundantly essential for the suppression of iNOS synthesis in macrophages by regulating IRF3 and STAT1 Pathways. Int Immunol. 2012;24:253-265.
58. Kuhr FK, Zhang Y, Brovkovych V, Skidgel RA. Betaarrestin 2 is required for B1 receptor-dependent posttranslational activation of inducible nitric oxide synthase. FASEB J. 2010;24:2475-2483.
59. Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3:339-345.
60. Meshki J, Douglas SD, Hu M, Leeman SE, Tuluc F. Substance P induces rapid and transient membrane blebbing in U373MG cells in a p21-activated kinasedependent manner. PLoS One. 2011;6:e25332.
61. Kajimoto H, Hashimoto K, Bonnet SN, Haromy A, Harry G, Moudgil R, et al. Oxygen activates the Rho/Rhokinase pathway and induces RhoB and ROCK-1 expression in human and rabbit ductus arteriosus by increasing mitochondria-derived reactive oxygen species: a newly recognized mechanism for sustaining ductal constriction. Circulation. 2007;115:1777-1788.
62. El-Remessy AB, Tawfik HE, Matragoon S, Pillai B, Caldwell RB, Caldwell RW. Peroxynitrite mediates diabetesinduced endothelial dysfunction: possible role of Rho kinase activation. Exp Diabetes Res. 2010;2010:247861.
63. McConalogue K, Déry O, Lovett M, Wong H, Walsh JH, Grady EF, et al. Substance P-induced trafficking of beta-arrestins. The role of beta-arrestins in endocytosis of the neurokinin-1 receptor. J. Biol. Chem. 1999;274:16257- 16268.
64. Teng X, Li D, Catravas JD, Johns RA. C/EBPbeta Mediates iNOS Induction by Hypoxia in Rat Pulmonary Microvascular Smooth Muscle Cells. Circ Res. 2002;90:125-127.
65. Izzi L, Attisano L. Regulation of the TGFbeta signalling pathway by ubiquitin-mediated degradation. Oncogene. 2004;23:2071-2078.
66. Neo S, Itahana Y, Alagu J, Kitagawa M, Guo AK, Lee S, et al. TRIM28 Is an E3 Ligase for ARF-Mediated NPM1/B23 SUMOylation That Represses Centrosome Amplification. Mol Cell Biol. 2015;35:2851-2863.
67. Li Y, Luo H, Liu T, Zacksenhaus E, Ben-David Y. The Ets Transcription Factor Fli-1 in Development, Cancer and Disease. Oncogene. 2015;34:2022-2031.
68. Truong AHL, Cervi D, Lee J, Ben-David Y. Direct transcriptional regulation of MDM2 by Fli-1. Oncogene. 2005;24:962-969.
69. Cheng Q, Cross B, Li B, Chen L, Li Z, Chen J. Regulation of MDM2 E3 ligase activity by phosphorylation after DNA damage. Mol Cell Biol. 2011;31:4951-4963.