Keywords
Aldehyde dehydrogenase, Stem cells marker, Transcriptional control, Retinoic acid
Abbreviations
ABC: ATP-Binding Cassette; ALDH1 A1/3: Aldehyde Dehydrogenases 1 isoforms A1, A3; AR: Androgen Receptor; C/EBPβ: CCAAT/Enhancer Binding Protein β; CHOP/ GADD153: Growth Arrest- and DNA Damage-inducible gene 153; CRABP2: Cellular Retinoic Acid Binding Protein 2; CRC: Colorectal Cancer; DDIT3: DNA Damage-Inducible Transcript 3; EMT: Epithelial-Mesenchymal Transition; ER: Estrogen Receptor; ERK: Extracellular signal-regulated Kinase; HGF: Hepatocyte Growth Factor; HOX: Homeobox gene; HSCs: Hematopoietic Stem Cells; KDM4C: Lysine histone demethylase 4C; MEK: Mitogen-activated protein Kinase; mTOR: Mammalian Target of Rapamycin; MUC1-C: Mucin 1 glycoprotein, subunit C; Nestin: Neuroepithelial stem cell protein; PI3K: Phosphatidylinositol 3-kinase; PPARβ/δ: Peroxisome-Proliferator-Activated Receptor β/δ; PRMT3: Protein Arginine Methyltransferase 3; RA: Retinoic Acid; RAR: Retinoic Acid Receptor; ROS: Reactive Oxygen Species; RXR: Retinoid X Receptor; SHH: Sonic Hedgehog; SNAI2: (Slug) zinc-fingered protein 2; SOX2: SRY (sex determining region Y)- box 2; TCF: T-cell Factor; TGF-β: Transforming Growth Factor β; ZEB1: Zinc finger E-box Binding homeobox 1
Introduction
The ALDH gene superfamily encodes a group of evolutionarily-related proteins catalyzing the irreversible oxidation of aldehyde substrates to their corresponding carboxylic acids. Aldehyde dehydrogenases (ALDH) isoforms 1A1 and 1A3 belong to intracellular enzymes with a broad spectrum of functions linked with an advanced stage of solid tumors and the stemness potential of the neoplastic cells. ALDH1A1 and ALDH1A3 share the ability to detoxicate aldehyde and regulate tumor initiation and progression by triggering retinoic acid (RA) signaling. In the focal article [1], we compiled the molecular mechanism of ALDH1A1 and ALDH1A3 transcription and regulation and their relation to the main molecular pathways responsible for the tumor cells’ excessive proliferation, chemoresistance, and stem cell properties.
This commentary would like to contribute to the abovementioned knowledge and report new findings confirming the metastatic potential of ALDH1A3, a discovery of new molecules connected to ALDH1A1/1A3 isoforms, and a phenomenon of genetic compensation regarding ALDH isoforms.
Transcriptional Regulation of ALDH1A1 and ALDH1A3 in Tumors
The aberrant expression of ALDH1A1 and ALDH1A3 isoforms is tissue-dependent and regulated transcriptionally by several signaling pathways (Figure 1) as already described in the focal review [1]. Briefly, ALDH1A1 is activated by the Wnt pathway via β-catenin and T-cell factor in breast cancer and prostate cancer [2]. β-catenin can be activated also by a Wnt-independent mechanism, downstream of the Akt axis, as reported in ALDHhigh cervical cancer cells [3]. Transforming growth factor β (TGF- β) pathway results in a downregulation of ALDH1A1 through Smad4 in cholecarcinoma [4], breast cancer [5], pancreatic cancer [6], and uterine endometrioid cancer [7]. Additionally, in breast cancer, mucine subunit MUC1-C can form a complex with the C/EBPβ transcription factor and activates the ALDH1A1 promotor. Epigenetically, ALDH1A1 activity can be reduced by acetylation, and the ALDH1A1 promoter is suppressed by chromatin looping with super-enhancer domain in presence of BRD4 protein in ovarian cancer cells [8].
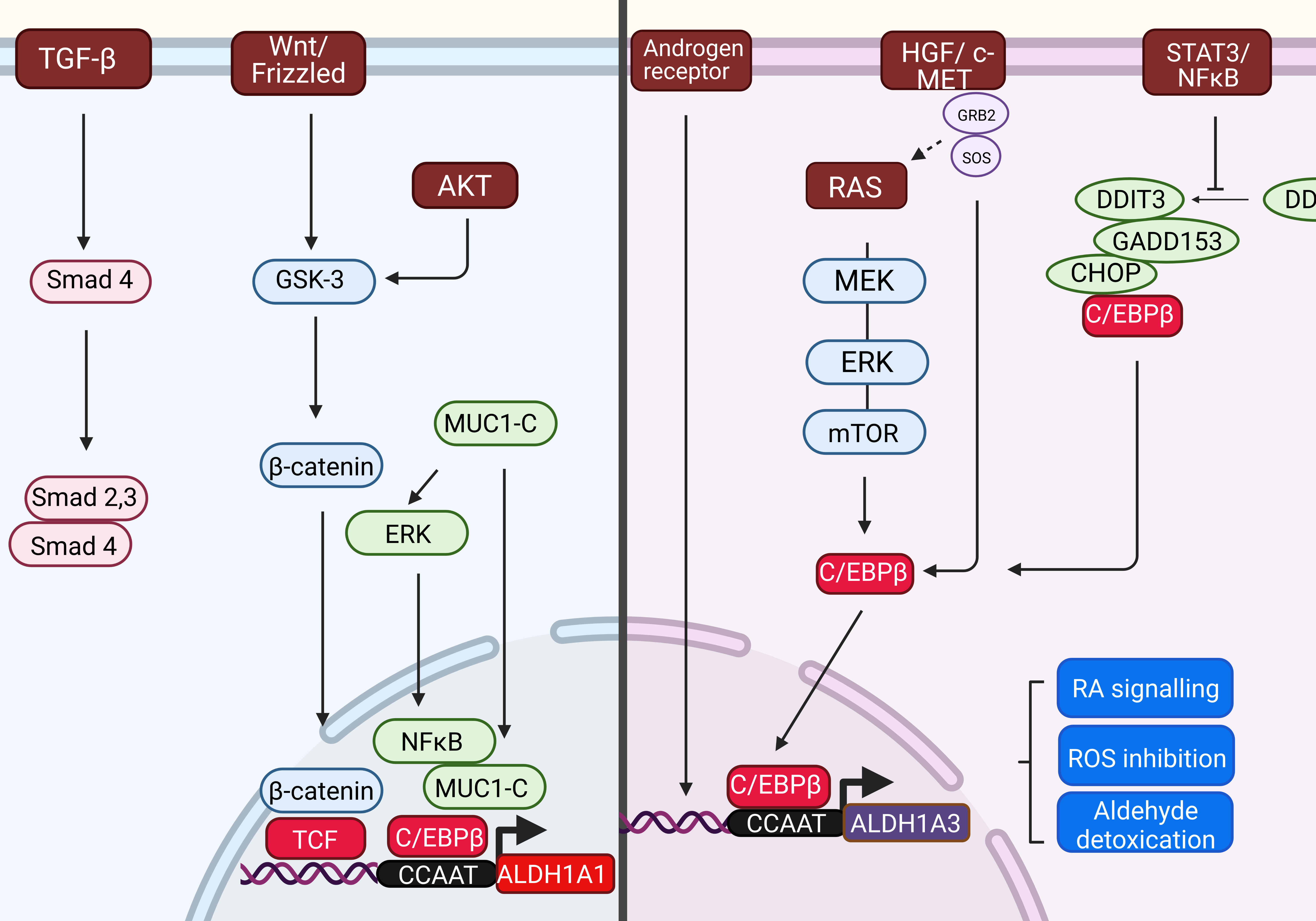
Figure 1: Transcriptional regulation of ALDH1A1 and ALDH1A3 genes. The upstream signaling pathways, which triggers the expression of ALDH1A1 gene are different to those regulating the expression of ALDH1A3 gene. Both the human ALDH1A1 and ALDH1A3 gene promoters contains a CCAAT box (71 to 67 bp) that is activated by C/EBPβ.
ALDH1A1 promoter can be activated by several molecular pathways: 1) Wnt pathway regulating ALDH1A1 through β-catenin and T-cell factor (TCF)-dependent transcription; 2) Transforming growth factor β (TGF-β) - downregulating ALDH1A1 in Smad4-dependent manner; 3) Mucin 1-C (MUC1-C) inducing extracellular-signal-regulated kinase (ERK) signaling and phosphorylating CCAAT enhancer binding protein (C/EBPβ), which leads to induction of ALDH1A1 expression.
ALDH1A3 expression can be activated by 1) the K-RAS/MEK/ERK/mTOR pathway; 2) Androgen Receptor in androgen-dependent cancer; 3) HGF/c-MET pathway; 4) STAT3-NFκB pathway, which activity represses the forming of complex with DDIT3/CHOP/GADD153 leading to CEBPβ-dependent ALDH1A3 promoter activation.
Abbreviations: AR: Androgen Receptor; C/EBPβ: CCAAT/Enhancer Binding Protein β; ERK: Extracellular signal-regulated Kinase; MEK: Mitogen-activated protein Kinase; mTOR: Mammalian Target of Rapamycin; HGF: Hepatocyte Growth Factor; DDIT3: DNA Damage Inducible Transcript 3; RA: Retinoic Acid; DHT: Dihydrotestosterone; CHOP/GADD153: Growth Arrest- and DNA Damage-inducible gene 153; DDIT: DNA-Damage Inducible transcript 3; MUC1-C: Mucin 1 subunit C; TCF - T-cell Factor; TGF-β: Transforming Growth Factor β.
The ALDH1A3 transcription was triggered by (i) KRAS oncogene and KRAS/MEK/mTOR pathway in pancreatic ductal adenocarcinoma [9], (ii) by HGF/ c-MET pathway in pancreatic cancer [10], (iii) Androgene receptor in human prostate cancer cell line [11] and by (iv) Signal transducer and activator of transcription 3 (STAT3)/ NFκB axis in pleural mesothelioma chemoresistant cell subpopulations [12]. Moreover, ALDH1A3 aberrant expression can be regulated by the tumor suppressor TP53 and epigenetically through promotor methylation [13] and by autophagy [1,14].
Although the activating pathways are different for ALDH1A1 and ALDH1A3 isoforms, a promotor region of both genes obtains a CCAAT region, which is recognized and bound by CCAAT/enhancer- binding protein β (C/EBPβ).
Recent data performed on thyroid cancer cell lines and patient tissue samples revealed that the transcriptional levels of ALDH1A1 and ALDH1B1 significantly decreased in thyroid cancer tissues vs. normal thyroid, whereas that of ALDH1A3 increased [15]. Their study of the protein-protein interactions revealed that ALDH1A1 can interact with transcription factors C/EBPβ and membrane-bound proteins ATP-binding cassette subfamily C member 6 (ABCC6) and mucin 1 (MUC1) [15].
ALDH1A1 and ALDH1A3 as Molecular Players in Tumor Progression
Both ALDH1A1 and 1A3 isoforms are involved in plenty of cellular processes but share many common functionalities. They participate in the cellular functions essential for cell survival along with cell protection. Their major function is irreversible oxidation of the endogenous and exogenous aldehydes to corresponding acids in presence of NAD+. They participate in physiological processes such as (i) Glycolysis/ Gluconeogenesis in converting acetaldehyde to acetic acid; (ii) Synthesis of amino acids histidine, β-alanine, phenylalanine, and tyrosine; (iii) Lipid peroxidation metabolizing products as hexanal, octanal, and others to the corresponding acids; (iv) Detoxification of exogenous aldehydes via cytochrome P450; (v) Reduction of intracellular oxidative species to defend the cell against oxidative damage; and the most important (vi) Conversion of retinol to retinoic acid (Figure 2). Subsequently, retinoic acid combines with cellular retinoic acid binding protein 2 (CRABP2) in target tissue cells and activates retinoic acid receptor (RAR) and retinoid X receptor (RXR) (or ER or PPARβ/δ, respectively) to trigger the transcription of RAresponsive genes, regulating proliferation, differentiation, cell cycle arrest and apoptosis [13]. RARs bind target genes as a heterodimer complex with RXRx at a DNA sequence known as the RA response element (RARE). RARE binding recruits either nuclear receptor coactivators or corepressors, thus directly activating or repressing transcription [16].
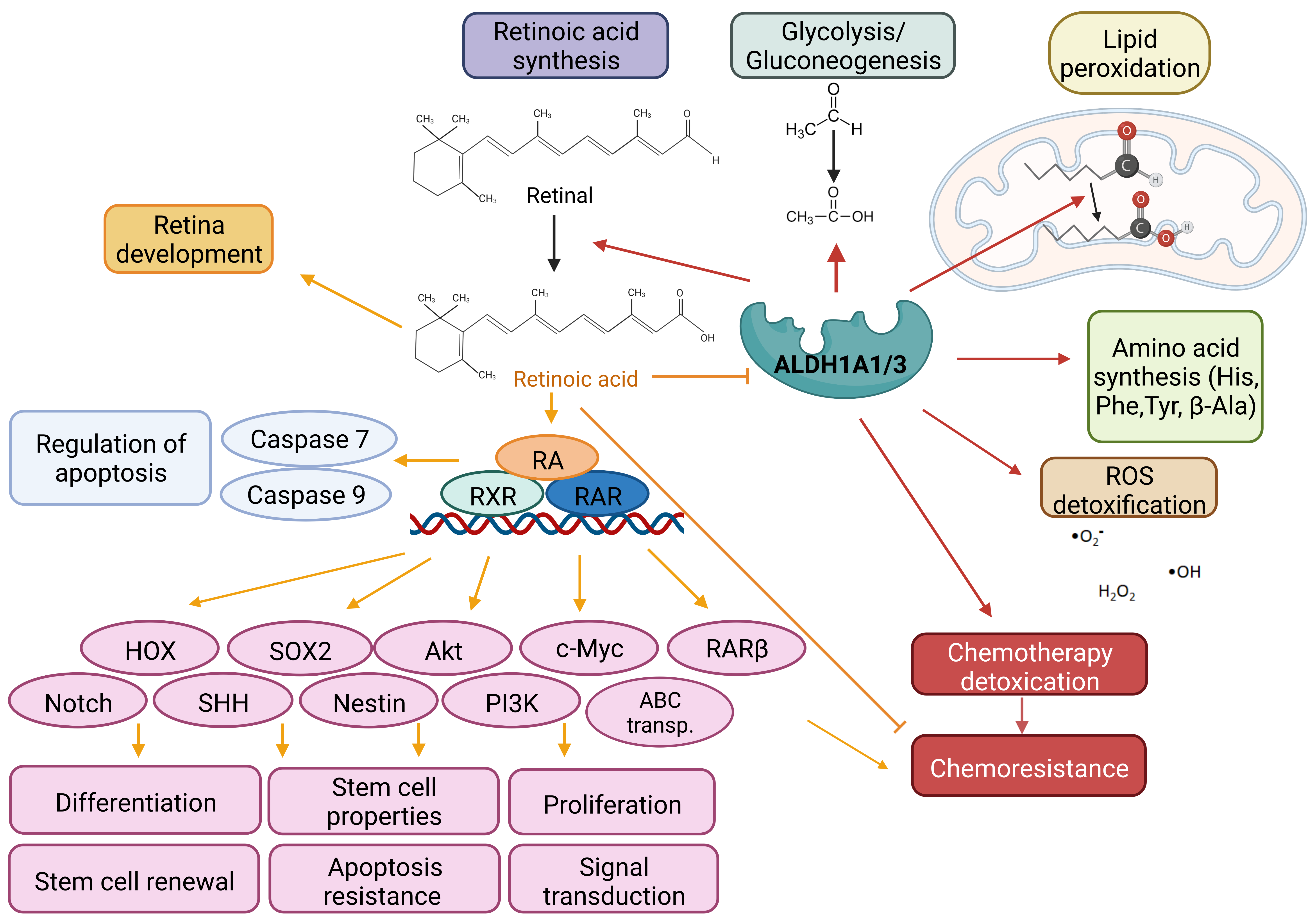
Figure 2: Mechanism of action of ALDH1A1 and 1A3 isoforms and retinoid acid. Main function of ALDH1A1 and 1A3 isoforms is irreversible oxidation of aldehyde to corresponding acids. Both isoforms are essential for the cell metabolism, survival and protection, because they are involving in: (1) Glycolysis/Gluconeogenesis; (2) Synthesis of amino acids; (3) Detoxication of exogenous and endogenous aldehydes; (4) Reduction of intracellular ROS to prevent an oxidative damage; and (5) Conversion of retinol to retinoic acid. Retinoic acid activates RAR and RXR in target tissue resulting in the transcription of RA- responsive genes, regulating proliferation, differentiation, stem cell properties, cell cycle arrest and apoptosis, and chemoresistance.
Abbreviations: ABC: ATP-Binding Cassette; HOX: Homeobox gene; Nestin: Neuroepithelial stem cell protein; PI3K: Phosphatidylinositol 3-Kinase; RAR: Retinoic Acid Receptor; RXR: Retinoid X Receptor; ROS: Reactive Oxygen Species; SHH: Sonic Hedgehog; SOX2: SRY (sex determining region Y)-box 2.
Cancer cells with aberrant expression of ALDH1A1/3 abuse their functions to defend the cell against oxidative stress and cytotoxic aldehydes generated during chemotherapy treatment leading to chemo- and radioresistance. Both ALDH1A1 and 1A3 isoforms play a regulatory role in the initiation and progression of tumors through the aldehyde clearance and the production of RA. RA induces the production of various genes that contribute to excessive proliferation (Akt, c-Myc, PI3K), changes in differentiation and signal transduction (Notch, Sonic Hedgehog, HOX), stem cell properties (SOX2, Nestin), interventions into the cell cycle and regulation of apoptosis (Caspase 7 and 9) and chemoresistance (ABC transporters) (Figure 2) [17]. Increased ALDH1A1 expression correlates with increased cell proliferation, colony/sphere formation ability, and resistance to common chemotherapy regimens in vitro and with tumorigenicity in vivo [18]. ALDH1A3 expression is cancer-specific and is connected also with colony/sphere forming, angiogenic activity in vitro, and tumorigenesis in vivo [13].
ALDH1A3 is Associated with Metastatic Potential in Colorectal Cancer
Previously the role of ALDH1A3 on migratory ability and metastatic potential was confusing. It has been stated that ALDH1A3 has a dual role in breast cancer metastatic promotion [19]. In colorectal cancer (CRC), ALDH1A3 appeared to be an indicator of the invasive and metastatic properties of colorectal cells [20]. In concordance with this, Duan's team [21] reported that ALDH1A3 overexpression in CRC cell line HCT116 and primary CRC1 cells triggered in vitro spontaneous invasive potential, and ALDH1A3 silencing reduced it. Additionally, ALDH1A3 overexpression significantly enhanced the metastatic potential of HCT116 cells in a mouse orthotopic xenograft model, and ALDH1A3 knockdown inhibited its metastatic capacity. Moreover, co-interference in HCT116 cells confirmed that ALDH1A3 supported the epithelialmesenchymal transition (EMT) of colorectal cancer via ZEB1 and SNAI2 transcriptional factors. The regulation of ZEB1 and SNAI2 expression was not direct, but ALDH1A3 regulated the mRNA stability of ZEB1 and SNAI2 via miR-200s. Following it, the treatment with the miRNA-200b repressor inhibited the epithelial phenotype induced by ALDH1A3 knockdown. Thus, miR-200 family members are involved in the ALDH1A3-mediated EMT in colorectal cancer by targeting ZEB1 and SNAI2 [21].
Novel Molecules Interacting with ALDH1A1 and 1A3 Isoforms
Interactions among ALDH1A1 and genes responsible for cell differentiation, multidrug resistance, redox balance, Polycomb, and Hox genes contribute to the excessive proliferation, tumorigenesis, chemoresistance, and stemness of cancer cells (reviewed in [1]). Recently, novel discoveries about the connection of ALDH1A1 to the Protein arginine methyltransferase (PRMT3) and ALDH1A3 connection to the Lysine histone demethylase 4C (KDM4C) were published.
KDM4C belongs to the Jumonji domain-2 family of histone demethylases and acts as an oncogene in oesophageal, gastric, prostate, breast [22], head, and neck [23], and other cancer. Dysregulation of KDM4C causes epigenetic changes in gene expression associated with tumorigenesis. In prostate cancer, KDM4C directly binds to the promoter of the c-Myc gene and triggers the expression of the c-Myc oncogene [22]. In gastric cancer, KDM4C regulates the transcription of ALDH1A3 by binding to the ALDH1A3 promoter and inhibits H3K9me2 (H3 histone methylated on 9th Lysine) level at the ALDH1A3 promoter. KDM4C epigenetically activates ALDH1A3 transcription through histone demethylation, and this process is critical for KDM4C-induced gastric cancer stemness maintenance [24]. Moreover, Lang's team revealed that the activated ALDH1A3 in turn, transcriptionally activates KDM4C by RA signaling, so there is the feed-forward loop supporting the stemness, tumorigenesis, and chemoresistance of gastric cancer. Genetic attenuation of KDM4C and pharmacological inhibition of ALDH1A3 sensitized the sphere-derived gastric cells to 5-fluorouracil and cisplatin both in vitro and in vivo [24].
ALDH1A1 was identified as an interaction partner of Protein arginine methyltransferase 3. PRMT3 regulates protein functions by catalyzing asymmetric demethylation of the arginine residues in proteins. It was confirmed the catalytic domain of PRMT3 interacts with the C-terminal region of ALDH1A1 [25]. PRMT3 does not methylate ALDH1A1 but directly binds to ALDH1A1, inhibiting its specific enzymatic activity and the expression of retinoic acid-responsive genes. Surprisingly, PRMT3 inhibits the enzymatic activity of ALDH1A1 without affecting the abundance of ALDH1A1. This opened the question about the link between the ALDH1A1 enzymatic activity and the level of ALDH1A1. Verma et al. detected the upregulation of ALDH1A1 in lung cancer; besides a significant downregulation of RA-responsive genes, possibly owing to the elevated levels of PRMT3. They implied that ALDH1A1 activity might not be a linear function of its abundance and rather an ALDH1A1 isoform activity than its levels might be considered for cancer prognosis [25]. However, other data supporting this theory were not published.
Aspects of the Genetic Compensation Regarding ALDH Isoforms
Generally, organisms have evolved an ability to maintain their living conditions despite minor differences in the genetic equipment, called genetic robustness [26]. Genetic robustness can manifest as a result of the loss of function of a specific protein. To manage it the organism has evolved redundant genetic pathways for carrying out essential cellular processes. The lost gene and its substitute can be by non-homologous gene pair if they encode parallel pathways that moderate a shared cellular process [27]. Upregulation of related genes following a gene knockout may be a direct consequence of the loss of protein function, which is called genetic compensation [26]. We can hypothesize that genetic compensation can be the way the tumor cells survive after the loss of the vital stem cell marker.
ALDH1A1 and ALDH1A3 are evolutionarily related, close in the phylogenetic tree, and both able to detoxicate aldehyde and regulate the tumor initiation and progression by triggering the retinoic acid signaling. However, ALDH1A1 is much less efficient at generating all-trans RA than ALDH1A2 and ALDH1A3. A question has been raised if there is a possibility of genetic compensation for the loss of one ALDH isoform by the rise of the expression of other ALDH isoform/s.
Some authors tested this possibility in mouse hematopoietic stem cells (HSCs), because Aldh1a1 and Aldh3a1 are both critical for HSC biology. Gasparetto et al. prepared a mouse knockout of Aldh1a1, but it did not affect HSCs or haematopoiesis. However, Aldh1a1 deficiency was associated with increased expression of the Aldh3a1 isoform. They reported that Aldh3a1 has the potential to compensate for Aldh1a1 in the bone marrow and is essential in B-cell development [28].
Both ALDH1A1 and ALDH1A3 regulate retinoic acid (RA) biosynthesis. Retinoic acid produced in the dorsal retina is essential for mammalian ocular development in a paracrine fashion. In the mouse system, Aldh1a1-null mice are born ordinarily with no developmental ocular phenotype [29], likely due to compensation in RA-signaling by ALDH1A3. Aldh1a3- null mice display all symptoms of developmental ocular phenotypes. Double knockout mice with Aldh1a1/Aldh1a3- null genotype exhibit identical phenotypes as Aldh1a3-null mice but with greater severity. This may indicate that the loss of RA-signaling induced by the genetic ablation of Aldh1a3 is partly compensated by Aldh1a1 [29, 30].
ALDH1A1 is also strongly expressed in the male somatic cells of the developing mouse testis. Surprisingly, Aldh1a1-null mice and Aldh1a1/Aldh1a3 double-knockout mice showed no obvious gonadal or fertility defects [31]. Aldh1a3, Aldh1a2, and Aldh1a8 are expressed at only low levels and not sexspecifically, suggesting that they may not compensate for Aldh1a1 in mouse gonads. Aldh1a1 (NM_013467) is almost identical to the Aldh1a7 sequence (NM_011921) in mice, located on chromosome 19 with the transcripts encoded in opposite directions. Aldh1a7 was 12.6-fold more highly expressed in the male gonad than in the female. So Aldh1a7 could compensate functionally in the Aldh1a1 knockout unless RA production [31]. ALDH1A7 is not present in the human genome.
Interestingly, Aldh1a1 is overexpressed and massively expanded in the genome of North American beavers, protecting them against exposure to aldehydes from lipid oxidation. Although they investigated whether the expansion of beaver Aldh1a1 is a compensation for the loss of other Aldh genes or a result of natural selection, they detected no changes in other Aldh genes. Beavers have the same Aldh genes as mice and humans, with regular gene structures and open reading frames [32].
In human glioma stem cells, Li et al. analyzed the expression of ALDH1A1 and ALDH1A2 upon the loss of ALDH1A3 in the six GSC-326/ALDH1A3-KO single-cell clones compared to the three GSC-326 (ALDH1A3 WT) control cells by microarray analysis. They did not detect any significant change in the expression of ALDH1A1 or ALDH1A2 [33].
A recent study [34] using genetic inhibition of ALDH1A1 and ALDH1A3 via siRNA in prostate cancer cell lines displayed that genetic silencing of ALDH1A1 led to downregulation of ALDH1A3; however, the depletion of ALDH1A3 increased ALDH1A1 mRNA expression [34].
The above-mentioned results are tissue-dependent and contradictory. The loss of one gene may be compensated by another gene with overlapping functions and expression patterns, often a paralogue and it is less likely in tissue with a low endogenous level [35].
The molecular mechanism of the genetic compensation is not elucidated. The closest is the mechanism of the transcriptional adaptation response described by El-Brolosy and Stainier [26]. They presented the hypothesis that consequently, after DNA damage (e.g. caused by genetic editing), histone-modifying enzymes cause global chromatin reorganization, positively affecting chromatin accessibility around the compensating gene(s), thereby leading to increased expression levels and its/their upregulation [26]. Alternatively, a mutant transcript is targeted for degradation by the RNA surveillance machinery during translation. As RNA degradation sequences are similar to compensatory genes, RNA intermediates can (i) guide RNA binding proteins to regulatory regions of compensatory genes or (ii) bind to and deactivate antisense transcripts inhibiting a compensatory gene [26,35]. Several studies suggest that the trigger of genetic compensation is upstream of protein function in transcriptional regulation. Both ALDH1A1 and ALDH1A3 promoters contain a CCAAT box (71 to 67 bp) that could be similarly activated by C/EBPβ. ALDH1A1 can be transcriptionally induced by the MUC1-C pathway, leading to the ERK activation and activation of the C/EBPβ transcription factor, binding to the ALDH1A1 promotor. Activated C/EBPβ can also bind to the ALDH1A3 promotor as the only known mechanism of ALDH1A3 activation [1].
Genetic compensation has to be considered in the case of studies of new inhibitors. For example, in non-small cell lung cancer cell lines, transient knockdown of highly abundant ALDH1A1 in the H460 cell line and ALDH1A1 together with ALDH1A3 in the HCC827 cell line led to a compensatory increase in ALDH3A1 protein expression but did not significantly change cell viability. Only simultaneous transient depletion of ALDH1A1 and/or ALDH3A1 significantly reduced cell viability by 81.5% and depletion of ALDH1A3 and/or ALDH3A1 decreased viability by 70.5%. So, the use of dual inhibitors of both the ALDH1 and ALDH3 isoform group could be considered for treatment improvement in lung carcinomas [36].
Conclusion
Recent findings confirm the important role of ALDH1A1 and ALDH1A3 in cancer biology. Further investigation could elucidate ALDH1-associated mechanisms enabling survival and metastatic dissemination of tumor cells in detail and help propose innovative therapeutic strategies. Genetic compensation of ALDH1 isoform remains an unsolved puzzle and might be the potential mechanism for how the cancer cells gain the growth advantage or escape from apoptosis.
Funding
This study was supported by the VEGA grants No. 2/0170/22 and 2/0050/19, by The Ministry of Health of the Slovak Republic under the contract 2019/60-BMCSAV-4.
Conflict of Interest Statement
Authors declare no conflicts of interest.
Competing Interests
Authors declare no competing interest.
Author Contributions Statement
Both authors contribute to this paper with the conception of the study and literature review. MP prepared the figures. Both authors read and approved the final version of the manuscript.
References
2. Cojoc M, Peitzsch C, Kurth I, Trautmann F, Kunz-Schughart LA, Telegeev GD, et al. Aldehyde Dehydrogenase Is Regulated by β-Catenin/TCF and Promotes Radioresistance in Prostate Cancer Progenitor Cells. Cancer Res. 2015;75(7):1482–94.
3. Sarabia-Sánchez MÁ, Alvarado-Ortiz E, Toledo-Guzman ME, García-Carrancá A, Ortiz-Sánchez E. ALDHhigh Population Is Regulated by the AKT/β-Catenin Pathway in a Cervical Cancer Model. Front Oncol. 2020;10:1039.
4. Shuang ZY, Wu WC, Xu J, Lin G, Liu YC, Lao XM, et al. Transforming growth factor-β1-induced epithelial-mesenchymal transition generates ALDH-positive cells with stem cell properties in cholangiocarcinoma. Cancer Lett. 2014;354(2):320–8.
5. Bhola NE, Balko JM, Dugger TC, Kuba MG, Sánchez V, Sanders M, et al. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest. 2013;123(3):1348-58.
6. Hoshino Y, Nishida J, Katsuno Y, Koinuma D, Aoki T, Kokudo N, et al. Smad4 Decreases the Population of Pancreatic Cancer- Initiating Cells through Transcriptional Repression of ALDH1A1. Am J Pathol. 2015;185(5):1457-70.
7. Wang Y, Jiang Y, Tian T, Hori Y, Wada N, Ikeda J, et al. Inhibitory effect of Nodal on the expression of aldehyde dehydrogenase 1 in endometrioid adenocarcinoma of uterus. Biochem Biophys Res Commun. 2013;440(4):731-6.
8. Yokoyama Y, Zhu H, Lee JH, Kossenkov AV, Wu SY, Wickramasinghe JM. BET Inhibitors Suppress ALDH Activity by Targeting ALDH1A1 Super-Enhancer in Ovarian Cancer. Cancer Res. 2016;76(21):6320-30.
9. Kong B, Wu W, Cheng T, Schlitter AM, Qian C, Bruns P, et al. A subset of metastatic pancreatic ductal adenocarcinomas depends quantitatively on oncogenic Kras/Mek/Erk-induced hyperactive mTOR signalling. Gut. 2016;65(4),647-57.
10. Kim IG, Lee JH, Kim SY, Kim JY, Cho E. W. Fibulin-3 negatively regulates ALDH1 via c-MET suppression and increases γ-radiationinduced sensitivity in some pancreatic cancer cell lines. Biochem Biophys Res Commun. 2014;454(3),369-75.
11. Casanova-Salas I, Masiá E, Armiñán A, Calatrava A, Mancarella C, Rubio-Briones J, et al. MiR-187 Targets the Androgen-Regulated Gene ALDH1A3 in Prostate Cancer. PloS One. 2015;10(5):e0125576.
12. Canino C, Luo Y, Marcato P, Blandino G, Pass HI, Cioce M. A STAT3-NFkB/DDIT3/CEBPβ axis modulates ALDH1A3 expression in chemoresistant cell subpopulations. Oncotarget. 2015;6(14):12637-53.
13. Duan JJ, Yu JC, Xiu FG, Shi WB, Yu C. (2016). ALDH1A3, a metabolic target for cancer diagnosis and therapy. Int J Cancer. 2016;139(5):965-75.
14. Wu W, Schecker J, Würstle S, Schneider F, Schönfelder M, Schlegel J. Aldehyde dehydrogenase 1A3 (ALDH1A3) is regulated by autophagy in human glioblastoma cells. Cancer Lett. 2018;417:112-23.
15. Cui Y, Liu Y. Transcriptional Expressions of ALDH1A1/B1 as Independent Indicators for the Survival of Thyroid Cancer Patients. Front Oncol. 2022;12:821958.
16. Cunningham TJ, Brade T, Sandell LL, Lewandoski M, Trainor PA, Colas A, et al. Retinoic acid activity in undifferentiated neural progenitors is sufficient to fulfill its role in restricting Fgf8 expression for somitogenesis. PLoS One. 2015;10:e0137894.
17. Zanoni M, Bravaccini S, Fabbri F, Arienti C. Emerging Roles of Aldehyde Dehydrogenase Isoforms in Anti-cancer Therapy Resistance. Front Med. 2022;9:795762.
18. Fitzgerald TL, McCubrey JA. Pancreatic cancer stem cells: association with cell surface markers, prognosis, resistance, metastasis and treatment. Adv Biol Regul. 2014;56:45-50.
19. Marcato P, Dean CA, Liu RZ, Coyle KM, Bydoun M, Wallace M, et al. Aldehyde dehydrogenase 1A3 influences breast cancer progression via differential retinoic acid signaling. Mol Oncol. 2015; 9(1):17-31.
20. Durinikova E, Kozovska Z, Poturnajova M, Plava J, Cierna Z, Babelova A et al. ALDH1A3 upregulation and spontaneous metastasis formation is associated with acquired chemoresistance in colorectal cancer cells. BMC Cancer. 2018;18(1):848.
21. Duan JJ, Wang D, Cai J, Chen JJ, Zheng XX, Chen TQ, et al. An aldehyde dehydrogenase 1A3 inhibitor attenuates the metastasis of human colorectal cancer. Cancer Lett. 2022;536:215662.
22. Lin CY, Wang BJ, Chen BC, Tseng JC, Jiang SS, Tsai KK, et al. Histone Demethylase KDM4C Stimulates the Proliferation of Prostate Cancer Cells via Activation of AKT and c-Myc. Cancers. 2019;11(11):1785.
23. Dorna D, Paluszczak J. The Emerging Significance of Histone Lysine Demethylases as Prognostic Markers and Therapeutic Targets in Head and Neck Cancers. Cells. 2022;11(6):1023.
24. Lang T, Xu J, Zhou L, Zhang Z, Ma X, Gu J, et al. Disruption of KDM4C-ALDH1A3 feed-forward loop inhibits stemness, tumorigenesis and chemoresistance of gastric cancer stem cells. Signal Transduct Target Ther. 2021;6(1):336.
25. Verma M, Khan M, Kadumuri RV, Chakrapani B, Awasthi S, Mahesh A, et al. PRMT3 interacts with ALDH1A1 and regulates gene-expression by inhibiting retinoic acid signaling. Commun Biol. 2021;4(1):109.
26. El-Brolosy MA, Stainier D. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017;13(7):e1006780.
27. Innan H, Kondrashov F. The evolution of gene duplications: classifying and distinguishing between models. Nat Rev Genet. 2010;11:97-108.
28. Gasparetto M, Sekulovic S, Brocker C, Tang P, Zakaryan A, Xiang P, et al. Aldehyde dehydrogenases are regulators of hematopoietic stem cell numbers and B-cell development. Exp Hematol. 2012;40(4):318-29.
29. Matt N, Dupé V, Garnier JM, Dennefeld C, Chambon P, Mark M, et al. Retinoic acid-dependent eye morphogenesis is orchestrated by neural crest cells. Development. 2005;132(21):4789-4800.
30. Thompson B, Katsanis N, Apostolopoulos N, Thompson DC, Nebert DW, Vasiliou V. Genetics and functions of the retinoic acid pathway, with special emphasis on the eye. Hum Genomics. 2019;13(1):61.
31. Bowles J, Feng CW, Knight D, Smith CA, Roeszler, KN, Bagheri- Fam S et al. Male-specific expression of Aldh1a1 in mouse and chicken fetal testes: implications for retinoid balance in gonad development. Dev Dyn. 2009;238(8):2073-80.
32. Zhang Q, Tombline G, Ablaeva J, Zhang L, Zhou X, Smith Z, et al. Genomic expansion of Aldh1a1 protects beavers against high metabolic aldehydes from lipid oxidation. Cell Rep. 2021;37(6):109965.
33. Li J, Garavaglia S, Ye Z, Moretti A, Belyaeva OV, Beiser A, et al. A specific inhibitor of ALDH1A3 regulates retinoic acid biosynthesis in glioma stem cells. Commun Biol. 2021;4(1):1420.
34. Gorodetska I, Offermann A, Püschel J, Lukiyanchuk V, Gaete D, Kurzyukova A et al. The Distinct Role of ALDH1A1 and ALDH1A3 in the Regulation of Prostate Cancer Metastases. bioRxiv. 2021;05.08.443223.
35. Bunton-Stasyshyn R, Wells S, Teboul L. When all is not lost: considering genetic compensation in laboratory animals. Lab Anim. 2019;48:282-4.
36. Rebollido-Rios R, Venton G, Sánchez-Redondo S, Iglesias I, Felip C, Fournet G, et al. Dual disruption of aldehyde dehydrogenases 1 and 3 promotes functional changes in the glutathione redox system and enhances chemosensitivity in nonsmall cell lung cancer. Oncogene. 2020;39(13):2756–2771.