Abstract
Competent human DNA mismatch repair (MMR) corrects DNA polymerase mistakes made during cell replication to maintain complete DNA fidelity in daughter cells; faulty DNA MMR occurs in the setting of inflammation and neoplasia, creating base substitutions (e.g. point mutations) and frameshift mutations at DNA microsatellite sequences in progeny cells. Frameshift mutations at DNA microsatellite sequences are a detected biomarker termed microsatellite instability (MSI) for human disease, as this marker can prognosticate and determine therapeutic approaches for patients with cancer. There are two types of MSI: MSI-High (MSI-H), defined by frameshifts at mono- and di-nucleotide microsatellite sequences, and elevated microsatellite alterations at selected tetranucleotide repeats or EMAST, defined by frameshifts in di- and tetranucleotide microsatellite sequences but not mononucleotide sequences. Patients with colorectal cancers (CRCs) manifesting MSI-H demonstrate improved survival over patients without an MSI-H tumor, driven by the generation of immunogenic neoantigens caused by novel truncated proteins from genes whose sequences contain coding microsatellites; these patients’ tumors contain hundreds of somatic mutations, and show responsiveness to treatment with immune checkpoint inhibitors. Patients with CRCs manifesting EMAST demonstrate poor survival over patients without an EMAST tumor, and may be driven by a more dominant defect in double strand break repair attributed to the MMR protein MSH3 over its frameshift correcting function; these patients’ tumors often have a component of inflammation (and are also termed inflammationassociated microsatellite alterations) and show less somatic mutations and lack coding mononucleotide frameshift mutations that seem to generate the neoantigens seen in the majority of MSI-H tumors. Overall, both types of MSI are biomarkers that can prognosticate patients with CRC, can be tested for simultaneously in marker panels, and informs the approach to specific therapy including immunotherapy for their cancers.
Keywords
MSI, EMAST, Colorectal cancer, Neoantigens, Mismatch repair, DNA repair, Tumor immunology, Tumor microenvironment, Inflammation
Abbreviations
CRC: Colorectal Cancer; MSI: Microsatellite Instability; MSI-H: MSI-High; MSI-L: MSI-Low; EMAST: Elevated Microsatellite Alterations at Selected Tetranucleotide Repeats; IAMA: Inflammatory-associate Microsatellite Alterations; MSS: Microsatellite Stable; IL-6: Interleukin-6; 5-FU: 5-fluorouracil; MMR: Mismatch Repair; DSB: Double Strand Break; bp: Base pair
Introduction
DNA microsatellites are tandemly repeating short (1-6 base pairs) DNA motifs, such as mononucleotide (A)n, dinucleotide (CA)n, trinucleotide (CAG)n, and tetranucleotide (AAAG)n, with each motif potentially repeating up to 50 times. These short sequence DNA repeats are distributed ~100,000 times throughout the genome and account for approximately 3% of the human genome [1,2]. The vast majority of microsatellite sequences are found in non-coding DNA, with a small proportion of mono-, di-, and tri-nucleotide repeats occurring within exons that code for proteins. Mononucleotide repeats are the most frequent with [A/T]n repeats 300 times more common than [G/C]n repeats [3]. Among dinucleotide repeats, [GT/CA]n and [AT/TA]n classes are the most abundant whereas [GC/CG] is the least abundant class; in case of tetranucleotides, [AAAG/TTTC]n, [AAAT/TTTA]n, and [AAAC/TTTG]n are the most abundant classes [3]. Trinucleotide repeats are about three times less abundant than di- and tetranucleotides. Because of their repeat reiteration, mutation rates at microsatellites (often quoted ~10-4 to 10-3 nucleotides per locus per generation) is several orders of magnitude higher than that of other parts of genome, leading to their variability in length [4]. The hyper-variability in length coupled with a high degree of heterozygosity, presence of multiple alleles and rapid detection protocols made DNA microsatellites ideal polymorphic markers for genome mapping, population genetics, genetic linkage studies, forensics, and cancer diagnostics [4,5].
Although highly polymorphic between individuals, microsatellites remain stable in length and exhibit the same profile within all the cells of an individual, due to effective surveillance and correction of post-replication errors by an efficient DNA mismatch repair (MMR) system before mitosis [6,7]. When the MMR system loses its proficiency, hundreds to thousands of point mutations and microsatellite frameshifts (also known as microsatellite instability, or MSI) cause a hypermutated genotype due to an increase in the spontaneous mutation rate, ideal conditions for rapid neoplastic transformation and progression [8,9]. There are four human conditions which exemplify this genotype: (1) patients with Lynch syndrome, an inherited condition in which one of the MMR genes is mutated in the germline leading to development of colorectal cancer (CRC) and endometrial cancer at a young age and accounts for about 3% of all CRC cases; (2) patients with Constitutional Mismatch Repair Deficiency (CMMRD), an inherited condition in which two mutated MMR genes are found in the germline leading to extremely young ages of onset for cancer and accounts for <0.1% of all CRC cases; (3) patients with Lynch-like syndrome in which there is no inherited MMR mutation but CRCs contain two somatic MMR mutations and accounts for ~1% of all CRCs; and (4) patients with sporadic MSI-H CRCs due to somatic hypermethylation of the DNA MMR gene MLH1 and accounts for ~15% of all CRC cases [9-13]. In all of these 4 conditions, CRCs demonstrate MSIHigh (MSI-H), defined by the biochemical presence of mono- and/or di-nucleotide frameshifts, with at least 2 of 5 markers demonstrating instability [9-13].
There is a fifth MMR-defective condition that does not result in MSI-H and appears not to generate hypermutated CRCs. Elevated microsatellite alterations at selected tetranucleotide repeats or EMAST is defined by frameshifts in di- and tetranucleotide microsatellite sequences but not mononucleotide sequences [14,15]. EMAST is observed in 50% of CRCs, making it the most common MMR defect observed in humans. EMAST is generated by the presence of inflammation (and is also called inflammationassociated microsatellite alterations) as a somatic, nonmutation mechanism to inactivate MMR [14-19].
In this review, we highlight the role of DNA microsatellites and MMR deficiency in contributing to the cancer immunological landscape and subsequent patient cancer care, with a focus on CRC patients.
DNA Mismatch Repair and Mutational Behavior of Microsatellite Sequences
Slippage of DNA polymerase during DNA replication is believed to be major cause of microsatellite variation and therefore the slipped strand mispairing model is often used to describe the mutational process at DNA microsatellites [4,20]. During DNA replication, DNA polymerase pauses at repetitive sequences and transiently dissociates from the template strand. Subsequent incorrect reannealing of template and newly-synthesized strands generate unpaired nucleotide loops on either the newlysynthesized strand or the template strands, which results in addition (if loop formed on newly-synthesized strand) or deletion (if loop formed on template strand) of one or more of the repeat units of the microsatellite during the next round of DNA replication if left unrepaired (Figure 1A). MMR contributes to the fidelity of DNA synthesis by recognizing and correcting mispaired nucleotides and insertion/deletion lops (IDLs) during the S phase of DNA replication before mitosis to prevent intermediate mutations (heteroduplexes) from becoming permanent in dividing cells [7,14,21].
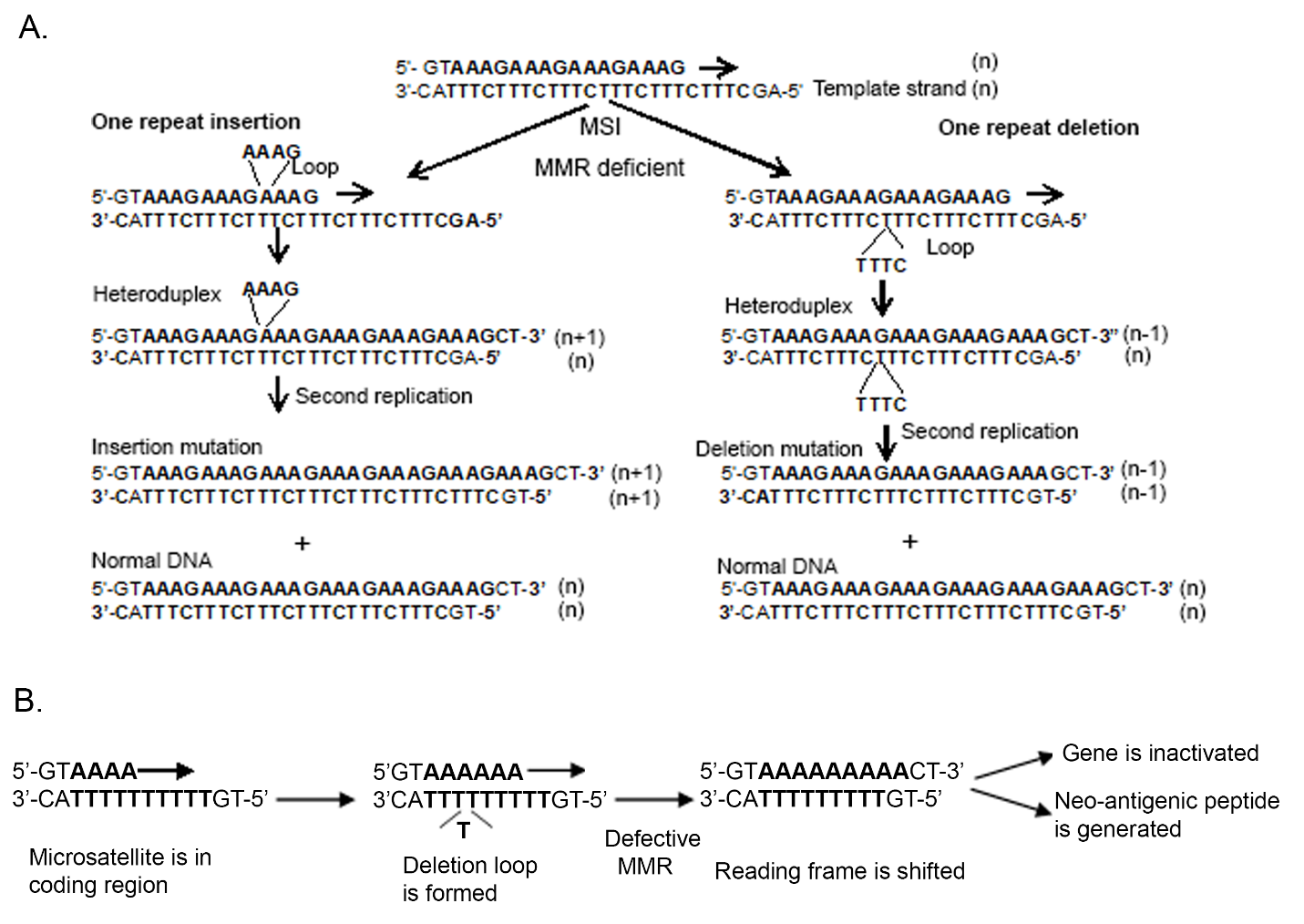
The highly-conserved heterodimeric protein complexes MutSα, MutSβ, and MutLα are the key players in MMRassociated genome maintenance [7,9,14]. MutSα and MutSβ play a critical role in mispair recognition in preparation for repair. MutSα (MSH2-MSH6 heterodimer) preferentially recognizes mismatched nucleotides and slippage errors at microsatellite loci with mono- and di-nucleotide repeats (generating insertion-deletion loops or IDLs of 1 to 2 bp), whereas MutSβ (MSH2-MSH3 heterodimer) recognizes slippage errors at microsatellite loci with di-, tri- and tetranucleotide repeats (IDLs of >2 bp) (Figure 2). Although, both complexes overlap in recognizing IDLs of 2 bp, MutSβ is more efficient in recognizing errors in dinucleotide repeats [22]. After recognition of the mismatch by MutSα or MutSβ, MutLα (MLH1-PMS2 heterodimer) is recruited to coordinate the excision of the mismatch, followed by DNA re-synthesis and re-ligation of the DNA strand. There are other MutL complexes including MutLβ (MLH1-PMS1 heterodimer) and MutLγ (MLH1-MLH3 heterodimer), but their role on post-replication DNA repair is not clear [7-9].
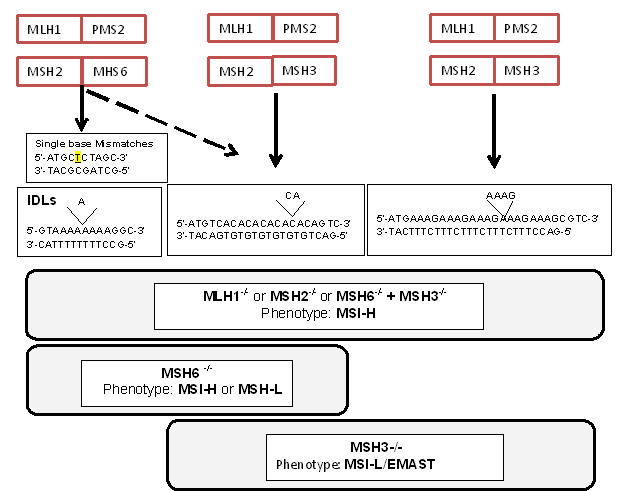
Figure 2. Human DNA MMR protein complexes, their preferences for different types of replication frameshift errors and resulting MSI phenotype in the absence of indicated MMR protein.
Loss of any component of MMR will cause frameshift mutations at microsatellites that will manifest as MSI (Figure 2). MSI models of cancer cells and mice show that loss of MLH1 or MSH2, the principle proteins in MMR complexes, completely abrogates MMR activity and results in genome-wide MSI at mono-, di-, tri- and tetranucleotides [7,8,15]. MSH2, the common heterodimer partner in both MutSα and MutSβ, is required for MSH6 and MSH3 protein stability and function within the nucleus, and likewise, loss of MLH1 results in degradation of PMS2 and therefore abrogates MutL activity [23]. In accordance with their essential functions, MSH2 and MLH1 are the most abundant proteins in the MMR system, and substantially more MutS proteins are present in comparison with the MutL proteins [23]. MSH2 primarily forms a heterodimer with MSH6 and therefore MutSα is more abundant in mammalian cell extracts compared to MutSβ. Imbalance in the relative amounts of the MSH3 and MSH6 proteins can result in MMR deficiency due to degradation of the partnerless protein [24]. MSH6 and MSH3 are considered partially redundant proteins and thus loss of function in either of these proteins generates a weaker mutator phenotype than that which occurs with loss of MLH1 or MSH2. For example, Loss of MSH6 affects only MutSα and generates mononucleotide and to a lesser extent dinucleotide MSI. Loss of MSH3 affects MutSβ function causing di-, tri-, and tetranucleotide MSI but not mononucleotide frameshifts (Figure 2) [25-28]. The partial redundancy between MutSα and MutSβ functions coupled with a compensatory increase in MSH3 expression and stability when MSH6 is absent indicates that most of the errors at dinucleotide repeats are repaired by MSH3 [23]. This genotype and function translate into phenotype: Msh3-/- or Msh6-/- mice develop tumors at a later age than Mlh1-/-or Msh2-/- mice [29] as do similar Lynch syndrome patients [10,12,26]. Combined Msh3-/- and Msh6-/- mice (double knockout) display a tumor disposition phenotype and high degree of microsatellite instability that are indistinguishable from those of Msh2-/- or Mlh1-/- mice, indicating that MSH3 cooperates with MSH6 in tumor suppression [29].
Mutation rates at microsatellites appear to be a complex phenomenon which is influenced by both polymerase slippage rate and MMR efficiency. It has been suggested that intrinsic factors specific to microsatellites, such as repeat number (length), motif size (mono-, di-, tri- , tetranucleotide), base composition of repeat motif, and complexity of microsatellite locus (pure repeats are less stable than mixed repeats) influence the primary slippage rate of microsatellites [2,4,30]. In addition, DNA sequences flanking the microsatellites can also modify polymerase slippage rate [2,30].
Both TGFBR2 (exon 3, A10) and ACVR2 (exon 10, A8) have coding polyadenine (mononucleotide) repeats. We previously measured the mutation rates of TGFBR2 and ACVR2 coding microsatellites in colon cancer cell lines with different MMR deficiencies (MLH1-/-, MSH6-/-, MSH3- /-, and MMR-proficient) using constructs that can measure frameshift mutation to place in-frame with EGFP [31]. We observed only 1-repeat deletion (i.e. A1) in the mutant population of both genes with MMR-deficiency, and mutation rates (mutations/cell/generation) for TGFBR2 and ACVR2 were dependent upon the human MMR background. MLH1 deficiency revealed eleven (5.91 x 10-4) and fifteen (2.18 x 10-4) times higher mutation rates for the TGFBR2 and ACVR2 microsatellites compared to MSH6 deficiency, respectively [31]. Importantly, no frameshift mutations were detected in MSH3-deficient and MMR proficient cells for both genes. Our data revealed that the coding microsatellite in TGFBR2 was more susceptible to mutation than ACVR2 in both MLH1- and MSH6- deficient cells. Moreover, ACVR2 contains two identical microsatellites in exon 3 and 10, but only the exon 10 microsatellite frameshifts in CRCs. To understand why the ACVR2 (A)8 microsatellite frameshifts in exon 10 but not in exon 3, we swapped a 6 bp DNA sequence flanking the 5’ and 3’ ends of the exon 3 and 10 microsatellites, and observed mutational behavior in MLH1-deficient cells [32]. The mutation rate of the exon 10 (A)8 tract was reduced 6-fold when its flanking sequence was swapped with exon 3, while substitution of flanking nucleotides from exon 10 to exon 3 enhanced frameshift mutation [32]. These results indicated that mutation selectivity for ACVR2 lie partly with flanking nucleotides surrounding each microsatellite. We also investigated the effect of microsatellite length upon frameshift mutation in gene-specific sequence context by varying the length of the (A)n repeats in TGFBR2 and ACVR2 [33]. Longer microsatellites showed significantly higher mutation rates within both genes; however, the rates of increase were different, suggesting that in addition to microsatellite length, the surrounding sequence context of microsatellite likely regulates frameshift mutation rate.
Finally, although our system was designed to detect only one-repeat (mononucleotide) deletion, the mutational spectra of the frameshifted A13 microsatellite within each gene showed more than one repeat deletion (33% were two-repeat deletions in TGFBR2) and 1-2 repeat insertions (8-10%), indicating that longer microsatellites are more mutable and continuously go through insertion/deletion mutations [33].
We utilized similar experiments to examine the mutational behavior of tetranucleotide sequences, which is the principle focus of EMAST [34]. Because we had observed the possibility of expansion as well as contraction at tetranucleotide sequences [28], which is different than the predominantly observed contraction at mononucleotide sequences [32], we created dual plasmid vectors containing various lengths of tetranucleotides repeats [(AAAG)12, (AAAG)15, (AAAG)18] surrounded by D9S242 flanking sequences (to maintain the same native mutability of all tested microsatellites), and microsatellites were inserted in EGFP reporter gene at +1 bp and -1 bp outof- frame positions [34]. Here, deletion and insertion of one or more AAAG repeat units due to slippage mutations would restore the reading frame of EGFP. We drew the following conclusions from our results: (1) tetranucleotide microsatellite mutation frequency increases with the length of the microsatellite, as observed with mononucleotide microsatellites [33], (2) tetranucleotide microsatellite mutation rates are linear as a function of microsatellite length within the same locus, (3) deletion mutation rates for longer tetranucleotide microsatellites are higher than insertion rates but equivalent between deletions and insertions for shorter tetranucleotide microsatellites, and (4) there is an accumulation of excessive deletion mutation events compared with insertion events by more than a 3-to-1 ratio over time for tetranucleotide microsatellites [34]. Our results suggest that insertions do occur, but often are converted into deletions through continual reversions that contract the number of tetranucleotide microsatellite units to give a broad diverse mutational spectrum in the absence of MMR. Although most of the alterations consisted of one or two repeats, repeat size were frameshifted to that between ≤ 10 to ≥ 20 for (AAAG)18 tracts within the accumulated mutations. The shorter the tetranucleotide microsatellite length, the more likely that a deletion was only 1-repeat unit. Overall, 78.9% of observed frameshifts were 1 AAAG repeat unit, 16.1% were 2 repeat units, and 5.1% were 3 or more repeat units [34]. A distinct mutational bias for deletions over time indicates that there is a preference for competent MMR to correct unpaired loops on the template DNA strand over unpaired loops on the newly-synthesized strand during DNA replication. This observation is highly important for development of combined MSI-H and EMAST marker panels, as tetranucleotide frameshifts can expand, then contract to its native length before shortening, and may appear not to have frameshifted in the absence of MMR function.
Generation of Neoantigens and Immune Response in MSI-H Colorectal Cancers
MSI-H tumors contain multiple mutations at coding and noncoding microsatellite sequences with variable frequencies leading to different functional consequences. There are multiple genes in human genome harboring short (7 to 10 repeat long) mononucleotide microsatellites in their coding regions and are mutational targets with absence of MMR activity [6,30]. Proteins encoded by these genes display tumor suppressive functions (regulators of growth, apoptosis, DNA repair) and deletion mutation in those mononucleotide tracks leads to a translational frameshift reading frame with subsequent loss of protein function due to truncation of the protein [35]. This novel protein becomes neo-antigenic to patient’s immune system (Figure 1B) [36,37]. The genes encoding for transforming growth factor-β receptor type II (TGFBR2), insulin-like growth factor II receptor (IGF2R), Bcl-2 associated X protein (BAX) and activin receptor type-2A (ACVR2) are among the most frequently mutated genes in MSI-H tumors [35,38,39]. Furthermore, several of the MMR genes (MSH6, MSH3, MLH3, PMS2, and MSH2) contain mononucleotide repeats in their coding region and thus can cause progressive and complete loss of all residual MMR activity once a major MMR gene, such as MLH1 and MSH2, is inactivated, and are believed to further accelerate genomic instability [35,40]. This increased rate of accumulation of mutations in genes responsible for regulating cell growth is the likely cause for rapid growth of adenomas and progression to carcinoma seen in Lynch syndrome and sporadic MSI-H CRCs [9,10]. Noncoding microsatellites, located in promoters, UTRs, and introns, can play important role in regulation of gene expression by affecting transcription rate, RNA stability, splicing, and RNA-protein interactions [41,42]. Deletion mutations of intronic mononucleotide repeats in ATM and MRE11 genes were found to result in protein-truncating aberrant splicing variants in MSI-H CRC tumors [43-45]. Mutations in the coding mononucleotide repeat of MBD4 can affect the approach to treatment of patients with MSI-H CRCs [46,47]. It’s likely that future research will identify more noncoding target sites in MSI-H CRCs.
The truncated neoantigens from the frameshifted coding regions are recognized by the immune system. This has been demonstrated by utilizing tumor-infiltrating T cells from MSI-H tumors and show that they recognize the frameshifted peptides [36,37]. T cells from the peripheral blood of sporadic MSI-H cancer patients and Lynch syndrome patients demonstrated response to the frameshifted peptides. Surprisingly, peripheral T cells from Lynch syndrome patients who did not have any developed cancer also demonstrated reactivity to the frameshifted peptides, suggesting tiny amounts of the neoantigens are being produced, immunizing the patient even before a cancer is formed [36].
The immune response to the frameshifted peptides is one that is protective to the patient, restraining tumor biological behavior (Figure 3). MSI-H CRCs show subepithelial lymphoid nodules, often referred to as a “Crohn’s like reaction” due to the similarity to granuloma formation in Crohn’s disease. There are increased tumor-infiltrating lymphocytes as compared to microsatellite stable (MSS) CRCs [9,16-18]. Patients with MSI- CRCs (both sporadic and Lynch) tend to present at an earlier TNM stage than their MSS counterparts, and likewise show longer survival [6,8,9,48]. Compared to patients with MSS CRCs, patients with MSI-H CRCs demonstrate increased activation of memory T cells, indicating a response to the generation of frameshifted peptides as neoantigens [49]. As MSI-H CRCs are hypermutated due to the lack of DNA MMR, tumors also acquire programmed cell death 1 (PD-1) receptor and/ or PD-1 ligand, making them more susceptible to immune checkpoint blockade (Figure 3) [49,50].
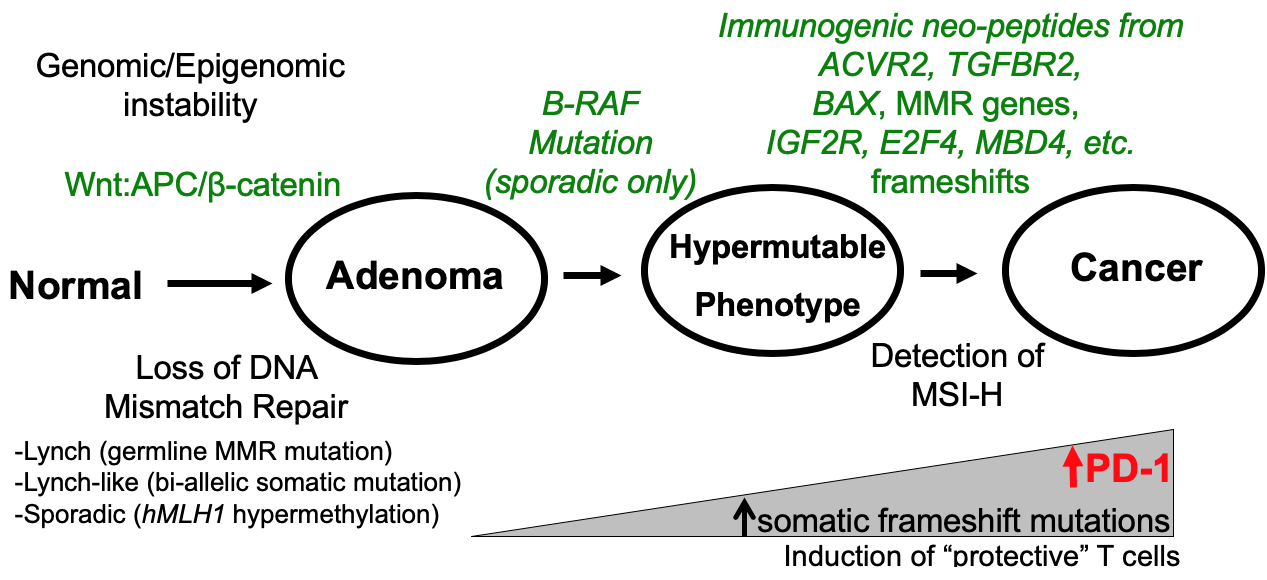
Figure 3: MMR Deficiency Pathogenesis in CRCs.
Most data for frameshifted peptide generation in the setting of MSI-H CRCs are generated by genes with coding mononucleotide microsatellites [9,36,37]. Thus, Lynch syndrome patients with MLH1, MSH2, MSH6, and PMS2 germline mutations, CMMRD patients, Lynch-like syndrome patients, and sporadic MSI-H CRC patients due to hypermethylation of MLH1 will generate frameshifted peptides. Patients with EMAST tumors (in which the sole defect is from MSH3 inactivation) would not be able to generate these mononucleotide frameshifted peptides, and lack the immune protection they afford after triggering the immune system.
EMAST Colorectal Cancers are a Consequence of Inflammation
Unlike MSI-H CRCs in which the DNA MMR defect occurs first to subsequently generate frameshifted peptides and elicits an immune response, EMAST CRCs are the result from the presence of innate inflammation. Inflammation and oxidative stress cause a reversible shift of MSH3 from the nucleus, where it is normally heterodimerized with MSH2 to effectuate DNA repair, to the cytosol where it no longer can repair nuclear DNA [28,51]. The proinflammatory cytokine that translocates MSH3 out of the nucleus is interleukin-6 (IL-6), which utilizes its JAK/ STAT3 intracellular pathway to relocate the protein [51]. MSH3 contains two cooperating nuclear export signals that transactivate IL-6’s effects, and has a single functional nuclear localization signal for eventual return to the nucleus once IL-6 has abated [52]. Thus, EMAST results from a somatic, non-mutational mechanism to inactivated DNA MMR. As a result of sole MSH3 loss of function, accumulation of di- and tetranucleotide frameshifts can proceed in the cell’s genome. Because inflammation initiates EMAST, this phenomenon can also be termed inflammation-associated microsatellite alterations (IAMA) and is observed in 50% of CRCs [19].
With the exclusion of other MMR defects, EMAST/IAMA CRCs are non-hypermutated; the combination of lack of a major MMR defect coupled with very few somatic mutations make these CRCs less likely to express PD-1/ PD-L1. Indeed, patients with EMAST CRCs show shorter survival due to possessing a more advanced tumor stage and more metastases as compared to patients with non- EMAST CRCs [14,48]. There is also a higher frequency of EMAST rectal cancers in African Americans compared to Caucasians, and a concomitant lessor frequency of MSI-H CRCs among African Americans [16,53]. In addition to the lack of immune response without the generation of mononucleotide coding frameshifts as seen in MSI-H CRCs, the absence of MSH3 function causes a second repair defect in double strand break (DSB) repair [14,54,55]. MSH3 (and the MMR complex MutSβ) participates in homologous recombination; in the absence of MSH3, accumulation of DSBs and aneuploidy ensue, tumor conditions that are associated with poor survival [8,9,56].
It should be noted that since EMAST/IAMA is initiated by inflammation, it can be observed in nonneoplastic tissue, possibly priming the tissue for future neoplastic transformation. EMAST is associated with benign hamartomatous polyps, can be identified in colonic adenomas, and is seen in ulcerative colitis with progression of the frequency of EMAST through dysplasia and colitis-associated CRC [17,18,57,58]. The common factor with these benign tissues and CRCs is the presence of inflammation driven by the pro-inflammatory cytokine IL-6.
Use of DNA Microsatellites for Molecular testing and Classification of MSI in tumors
Molecular testing for MSI is performed using PCR amplification of DNA regions containing microsatellite repeats, comparing lengths from normal and tumor tissue. Electrophoretic patterns of PCR products (detected as a mobility shift on electrophoretic gels) are then evaluated to identify insertions or deletions of repetitive units from the tumor sample as compared to normal. A standard panel of microsatellite markers was codified at a National Cancer Institute workshop in 1998, consisting of two mononucleotide repeat markers (BAT25 and BAT26) and three dinucleotide repeat markers (D2S123, D5S346, D17S250) [59]. MSI-H is defined as frameshift or instability in two or more of the five markers (or >30% of markers if more than five markers tested), and MSI-Low (MSI-L) is defined as an instability in one (or <30 of markers tested). Tumors are considered MSS when none of the markers tested show instability. Later, a revised Bethesda panel includes an additional three mononucleotide markers (NR- 21, NR-24, NR-27) to increase the screening sensitivity of tumors for MSI-H [60].
Detection of EMAST requires the use of tetranucleotide microsatellite markers that are not currently present in MSI-H panels. At present, there is no official consensus regarding the definition of EMAST, although most publications have used the definition of 1 or 2 frameshifted markers as positive from a panel that includes at least 5 or more various tetranucleotide markers (e.g. MYCL1, D9S242, D20S85, D8S321, D20S82, D19S394, RBM47, L17835, D2S443, D21S1436, D9S747, UTS037, L17686, UT5320, D11S488) [14,61].
Current microsatellite panels are designed to accurately identify MSI-H CRCs. MSI-L tumors typically show frameshifts at only one of three dinucleotide markers in the panel, and their morphological and clinical phenotype is similar to MSS tumors. Current evidence indicates that MSI-L classifies as EMAST, as both are caused by isolated loss of MSH3 function [14,27,28,48]. Combining these two subgroups as MSI-L/EMAST acknowledges that fact, and represents a group of patients demonstrating poor prognosis from CRC [48,56,62]. Accurate identification of the CRC type helps dictate the approach to treatment as well as prognosis for patients.
Immunotherapeutic Approach for Patients with CRC based on Microsatellite Alterations
Current MSI testing can identify patients who possess MSI-H CRCs and MSS CRCs. Identification of patients with EMAST CRCs, a subset of MSS CRCs, requires the use of tetranucleotide microsatellite markers, which have not yet made it to commercialized MSI panels. The designation of MSI-H and EMAST has treatment implications for those who require additional therapy beyond surgery.
Conventional chemotherapy regimens for advanced CRC treatment is based on the use of 5-fluorouracil (5- FU), typically with added leucovorin, irinotecan, and/ or oxaliplatin [63,64]. Patients with MSI-H CRCs are resistant to 5-FU because the MMR system, principally MutSα but also MutSβ, are needed to recognize 5-FU that gets incorporated into DNA (in addition to its effects on RNA) [65-67]. Lack of 5-FU recognition within DNA by MMR fails to trigger cell death, making the cancer cell resistant to 5-FU [65-67]. Most sporadic MSI-H CRCs as well as most Lynch and Lynch-like associated CRCs, due to inactivation of MLH1 or MSH2, are more resistant to 5-FU therapy [68,69]. In one study, MSI-H CRC patients treated with 5-FU did much worse with survival compared to untreated patients, possibly due to the inability to kill the MSI-H CRC cells while simultaneously killing the protective adaptive immune cells (which remain sensitive due to intact MMR) [69]. However, patients with EMAST/ IAMA CRCs, which lack MutSβ function but retain MutSα and MutLα function, remain sensitive to 5-FU (Table 1) [70].
| Type of MSI | Frequency among CRCs | Immune Mechanism | Patient prognosis (compared with MSS CRCs) | 5-fluorouracil sensitivity |
Immune Checkpoint Sensitivity |
| MSI-H | 15% | Generates protective adaptive inflammation |
Good | Low | High |
| EMAST/IAMA | 50% | Caused by innate inflammation |
Poor | Moderate to High | Low |
Immune checkpoint inhibitors such as anti-PD-1 are drugs that can remove a cancer cell’s self-preservation protection by blocking proteins that shield it from immune destruction. MSI-H tumors have at least 2 mechanisms that have shown them ideal to be responsive to immune checkpoint blockade. The first is by the nature of MSI-H tumors – its defect in MMR – make the tumor hypermutated, acquiring and coalescing hundreds to thousands of somatic mutations from the failed fidelity of DNA replication. Coding mononucleotide frameshifts generate neoantigenic peptides, inducing an adaptive immune response [9,36,37]. The second is the acquirement of immune checkpoint expression (e.g. PD-1) on the MSI-H cancer cell’s surface. Over 60% of CRCs with a high immunoscore (high levels of T cells at the invasive margin and center of the tumor) of which the majority are MSI-H, show PD-1 expression [49]. Clinical trials using anti-PD-1 demonstrated that patients with MSI-H tumors had significant biomarker and radiographic responses to the immune checkpoint therapy, resulting in 78% progression-free survival (PFS) vs 11% for patients with MSS tumors [50]. This and other data led the U.S. Food and Drug Administration to approve anti-PD-1 therapy for all tumors manifesting MSI-H, the first time ever that an oncological drug was approved for tumors across all body sites and solely based on the defect in DNA MMR [71,72]. Thus, even though MSI-H CRCs appear more resistant to 5-FU, patients with these tumors have an overall improved survival as compared to patients with MSS CRCs, and they can further gain oncological improvement with the use of immune checkpoint inhibitors (Table 1). Patients with EMAST/IAMA CRCs, whose tumors are nonhypermutated, would be expected to have less response to immune checkpoint inhibition.
Conclusion
MSI, the biochemical detection of frameshifted DNA microsatellite sequences, has become a powerful tool that can prognosticate CRC patients and can inform therapeutic approaches for their care. Presently, clinical MSI testing only assays frameshift mutations within mononucleotide and dinucleotide microsatellites which make up commercial panels, and can identify patients with MSI-H CRCs. MSI-H CRC patients show improved prognosis over MSS CRC patients as a result of the generation of neoantigenic peptides generated from frameshifted coding mononucleotide, providing an adaptive immune response that restrains the advancement of the tumor. This phenomenon, provided by a defect in MMR function that causes hundreds to thousands of somatic mutations, also enhances expression of PD-1/PD-L1, affording the opportunity to further improve patient outcome through the use of immune checkpoint inhibition. Thus, testing for MSI is critical in each and every CRC patient for these purposes.
Testing for EMAST/IAMA is not yet commercialized, but likely will be in the future. MSI panels would have to add tetranucleotide microsatellite markers to simultaneously distinguish MSI-H, MSS, and EMAST tumors. Patients with EMAST/IAMA CRCs show poor prognosis compared to their non-EMAST counterparts, remain sensitive to 5-FU therapy, and appear less sensitive to immune checkpoint inhibition likely because of lack of somatic hypermutation within the tumor genome. Since non-MSI-H CRC patients still receive 5-FU-based chemotherapy and are less eligible for immune checkpoint therapy as compared to MSI-H CRC patients, the clinical push to know a patient’s EMAST status is not as critical as knowing the MSI-H status; however, the EMAST status does provide prognostic information for the patient. Perhaps more importantly, EMAST is present in non-neoplastic inflammatory conditions that might project information on the ability for neoplastic transformation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest are disclosed.
Acknowledgements
Supported by the United States Public Health Service (R01 CA206010) and the A. Alfred Taubman Medical Research Institute of the University of Michigan. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Contributions
Both MOR and JMC equally contributed to all aspects of this manuscript.
References
2. Ellegren H. Microsatellites: simple sequences with complex evolution. Nature Reviews Genetics. 2004 Jun;5(6):435-45.
3. Subramanian S, Mishra RK, Singh L. Genomewide analysis of microsatellite repeats in humans: their abundance and density in specific genomic regions. Genome Biology. 2003 Feb 1;4(2):R13.
4. Ellegren H. Microsatellite mutations in the germline:: implications for evolutionary inference. Trends in Genetics. 2000 Dec 1;16(12):551-8.
5. Sidransky D. Nucleic acid-based methods for the detection of cancer. Science. 1997 Nov 7;278(5340):1054- 8.
6. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010 May 1;138(6):2073-87.
7. Carethers JM. Hereditary, sporadic and metastatic colorectal cancer are commonly driven by specific spectrums of defective DNA mismatch repair components. Transactions of the American Clinical and Climatological Association. 2016;127:81.
8. Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008 Oct 1;135(4):1079-99.
9. Carethers JM, Jung BH. Genetics and genetic biomarkers in sporadic colorectal cancer. Gastroenterology. 2015 Oct 1;149(5):1177-90.
10. Carethers JM, Stoffel EM. Lynch syndrome and Lynch syndrome mimics: The growing complex landscape of hereditary colon cancer. World Journal of Gastroenterology: WJG. 2015 Aug 21;21(31):9253.
11. Carethers JM. Differentiating Lynch-like from Lynch syndrome. Gastroenterology. 2014 Mar;146(3):602.
12. Stoffel EM, Carethers JM. Current Approaches to Germline Cancer Genetic Testing. Annual Review of Medicine. 2020 Jan 27;71:85-102.
13. Carethers JM. High predictability for identifying Lynch syndrome via microsatellite instability testing or immunohistochemistry in all Lynch-associated tumor types. Translational Cancer Research. 2019 Dec;8(Suppl 6):S559.
14. Carethers JM, Koi M, Tseng-Rogenski SS. EMAST is a form of microsatellite instability that is initiated by inflammation and modulates colorectal cancer progression. Genes. 2015 Jun;6(2):185-205.
15. Carethers JM. Microsatellite instability pathway and EMAST in colorectal cancer. Current Colorectal Cancer Reports. 2017 Feb 1;13(1):73-80.
16. Devaraj B, Lee A, Cabrera BL, Miyai K, Luo L, Ramamoorthy S, et al. Relationship of EMAST and microsatellite instability among patients with rectal cancer. Journal of Gastrointestinal Surgery. 2010 Oct 1;14(10):1521-8.
17. Lee SY, Chung H, Devaraj B, Iwaizumi M, Han HS, Hwang DY, et al. Microsatellite alterations at selected tetranucleotide repeats are associated with morphologies of colorectal neoplasias. Gastroenterology. 2010 Nov 1;139(5):1519-25.
18. Lee SY, Miyai K, Han HS, Hwang DY, Seong MK, Chung H, et al. Microsatellite instability, EMAST, and morphology associations with T cell infiltration in colorectal neoplasia. Digestive Diseases and Sciences. 2012 Jan 1;57(1):72-8.
19. Koi M, Tseng-Rogenski SS, Carethers JM. Inflammation-associated microsatellite alterations: Mechanisms and significance in the prognosis of patients with colorectal cancer. World Journal of Gastrointestinal Oncology. 2018 Jan 15;10(1):1.
20. Streisinger G, Okada Y, Emrich J, Newton J, Tsugita A, Terzaghi E, et al. Frameshift mutations and the genetic code. InCold Spring Harbor Symposia on Quantitative Biology 1966 Jan 1 (Vol. 31, pp. 77-84). Cold Spring Harbor Laboratory Press.
21. Carethers JM, Hawn MT, Chauhan DP, Luce MC, Marra G, Koi M, et al. Competency in mismatch repair prohibits clonal expansion of cancer cells treated with N-methyl-N’-nitro-N-nitrosoguanidine. The Journal of Clinical Investigation. 1996 Jul 1;98(1):199-206.
22. Kantelinen J, Kansikas M, Korhonen MK, Ollila S, Heinimann K, Kariola R, et al. MutSß exceeds MutSa in dinucleotide loop repair. British Journal of Cancer. 2010 Mar;102(6):1068-73.
23. Chang DK, Ricciardiello L, Goel A, Chang CL, Boland CR. Steady-state regulation of the human DNA mismatch repair system. Journal of Biological Chemistry. 2000 Jun 16;275(24):18424-31.
24. Marra G, Iaccarino I, Lettieri T, Roscilli G, Delmastro P, Jiricny J. Mismatch repair deficiency associated with overexpression of the MSH3 gene. Proceedings of the National Academy of Sciences. 1998 Jul 21;95(15):8568- 73.
25. Haugen AC, Goel A, Yamada K, Marra G, Nguyen TP, Nagasaka T, et al. Genetic instability caused by loss of MutS homologue 3 in human colorectal cancer. Cancer Research. 2008 Oct 15;68(20):8465-72.
26. Adam R, Spier I, Zhao B, Kloth M, Marquez J, Hinrichsen I, et al. Exome sequencing identifies biallelic MSH3 germline mutations as a recessive subtype of colorectal adenomatous polyposis. The American Journal of Human Genetics. 2016 Aug 4;99(2):337-51.
27. Campregher C, Schmid G, Ferk F, Knasmüller S, Khare V, Kortüm B, et al. MSH3-deficiency initiates EMAST without oncogenic transformation of human colon epithelial cells. PloS one. 2012 Nov 27;7(11):e50541.
28. Tseng-Rogenski SS, Chung H, Wilk MB, Zhang S, Iwaizumi M, Carethers JM. Oxidative stress induces nuclear-to-cytosol shift of hMSH3, a potential mechanism for EMAST in colorectal cancer cells. PloS one. 2012 Nov 30;7(11):e50616.
29. Edelmann W, Umar A, Yang K, Heyer J, Kucherlapati M, Lia M, et al. The DNA mismatch repair genes Msh3 and Msh6 cooperate in intestinal tumor suppression. Cancer Research. 2000 Feb 15;60(4):803-7.
30. Duval A, Hamelin R. Mutations at coding repeat sequences in mismatch repair-deficient human cancers: toward a new concept of target genes for instability. Cancer Research. 2002 May 1;62(9):2447-54.
31. Chung H, Young DJ, Lopez CG, Le TA, Lee JK, Ream- Robinson D, et al. Mutation rates of TGFBR2 and ACVR2 coding microsatellites in human cells with defective DNA mismatch repair. PLoS One. 2008 Oct 21;3(10):e3463.
32. Chung H, Lopez CG, Young DJ, Lai JF, Holmstrom J, Ream-Robinson D, et al. Flanking sequence specificity determines coding microsatellite heteroduplex and mutation rates with defective DNA mismatch repair (MMR). Oncogene. 2010 Apr;29(15):2172-80.
33. Chung H, Lopez CG, Holmstrom J, Young DJ, Lai JF, Ream-Robinson D, et al. Both microsatellite length and sequence context determine frameshift mutation rates in defective DNA mismatch repair. Human Molecular Genetics. 2010 Jul 1;19(13):2638-47.
34. Raeker MÖ, Pierre-Charles J, Carethers JM. Tetranucleotide Microsatellite Mutational Behavior Assessed in Real Time: Implications for Future Microsatellite Panels. Cellular and Molecular Gastroenterology and Hepatology. 2020 Jan 23.
35. Carethers JM, Pham TT. Mutations of transforming growth factor beta 1 type II receptor, BAX, and insulin-like growth factor II receptor genes in microsatellite unstable cell lines. In Vivo (Athens, Greece). 2000 Jan 1;14(1):13- 20.
36. Schwitalle Y, Kloor M, Eiermann S, Linnebacher M, Kienle P, Knaebel HP, et al. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology. 2008 Apr 1;134(4):988-97.
37. Kloor M, Michel S, von Knebel Doeberitz M. Immune evasion of microsatellite unstable colorectal cancers. International Journal of Cancer. 2010 Sep 1;127(5):1001- 10.
38. Jung B, Smith EJ, Doctolero RT, Gervaz P, Alonso JC, Miyai K, et al. Influence of target gene mutations on survival, stage and histology in sporadic microsatellite unstable colon cancers. International Journal of Cancer. 2006 May 15;118(10):2509-13.
39. Jung B, Doctolero RT, Tajima A, Nguyen AK, Keku T, Sandler RS, et al. Loss of activin receptor type 2 protein expression in microsatellite unstable colon cancers. Gastroenterology. 2004 Mar 1;126(3):64-59.
40. Perucho M. Microsatellite instability: the mutator that mutates the other mutator. Nature Medicine. 1996 Jun;2(6):630-1.
41. Li YC, Korol AB, Fahima T, Nevo E. Microsatellites within genes: structure, function, and evolution. Molecular Biology and Evolution. 2004 Jun;21(6):991-1007.
42. Hui J, Hung LH, Heiner M, Schreiner S, Neumüller N, Reither G, et al. Intronic CA-repeat and CA-rich elements: a new class of regulators of mammalian alternative splicing. The EMBO Journal. 2005 Jun 1;24(11):1988-98.
43. Ejima Y, Yang L, Sasaki MS. Aberrant splicing of the ATM gene associated with shortening of the intronic mononucleotide tract in human colon tumor cell lines: a novel mutation target of microsatellite instability. International Journal of Cancer. 2000 Apr 15;86(2):262- 8.
44. Giannini G, Ristori E, Cerignoli F, Rinaldi C, Zani M, Viel A, et al. Human MRE11 is inactivated in mismatch repair-deficient cancers. EMBO Reports. 2002 Mar 1;3(3):248-54.
45. Giannini G, Rinaldi C, Ristori E, Ambrosini MI, Cerignoli F, Viel A, et al. Mutations of an intronic repeat induce impaired MRE11 expression in primary human cancer with microsatellite instability. Oncogene. 2004 Apr;23(15):2640-7.
46. Suzuki S, Iwaizumi M, Tseng-Rogenski S, Hamaya Y, Miyajima H, Kanaoka S, et al. Production of truncated MBD4 protein by frameshift mutation in DNA mismatch repair-deficient cells enhances 5-fluorouracil sensitivity that is independent of hMLH1 status. Cancer Biology & Therapy. 2016 Jul 2;17(7):760-8.
47. Suzuki S, Iwaizumi M, Yamada H, Sugiyama T, Hamaya Y, Furuta T, et al. MBD4 frameshift mutation caused by DNA mismatch repair deficiency enhances cytotoxicity by trifluridine, an active antitumor agent of TAS-102, in colorectal cancer cells. Oncotarget. 2018 Feb 20;9(14):11477.
48. Garcia M, Choi C, Kim HR, Daoud Y, Toiyama Y, Takahashi M, et al. Association between recurrent metastasis from stage II and III primary colorectal tumors and moderate microsatellite instability. Gastroenterology. 2012 Jul 1;143(1):48-50.
49. Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D, et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 2016 Mar 15;44(3):698-711.
50. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatchrepair deficiency. New England Journal of Medicine. 2015 Jun 25;372(26):2509-20.
51. Tseng-Rogenski SS, Hamaya Y, Choi DY, Carethers JM. Interleukin 6 alters localization of hMSH3, leading to DNA mismatch repair defects in colorectal cancer cells. Gastroenterology. 2015 Mar 1;148(3):579-89.
52. Tseng-Rogenski SS, Munakata K, Choi DY, Martin PK, Mehta S, Koi M, et al. The human DNA mismatch repair protein MSH3 contains nuclear localization and export signals that enable nuclear-cytosolic shuttling in response to inflammation. Molecular and Cellular Biology. 2020 Jun 15;40(13):e00029-20.
53. Carethers JM, Murali B, Yang B, Doctolero RT, Tajima A, Basa R, et al. Influence of race on microsatellite instability and CD8+ T cell infiltration in colon cancer. PloS one. 2014 Jun 23;9(6):e100461.
54. Dietlein F, Thelen L, Jokic M, Jachimowicz RD, Ivan L, Knittel G, et al. A functional cancer genomics screen identifies a druggable synthetic lethal interaction between MSH3 and PRKDC. Cancer Discovery. 2014 May 1;4(5):592-605.
55. van Oers JM, Edwards Y, Chahwan R, Zhang W, Smith C, Pechuan X, et al. The MutSß complex is a modulator of p53-driven tumorigenesis through its functions in both DNA double-strand break repair and mismatch repair. Oncogene. 2014 Jul;33(30):3939-46.
56. Koi M, Garcia M, Choi C, Kim HR, Koike J, Hemmi H, et al. Microsatellite alterations with allelic loss at 9p24. 2 signify less-aggressive colorectal cancer metastasis. Gastroenterology. 2016 Apr 1;150(4):944-55.
57. Huang SC, Lee JK, Smith EJ, Doctolero RT, Tajima A, Beck SE, et al. Evidence for an hMSH3 defect in familial hamartomatous polyps. Cancer. 2011 Feb 1;117(3):492- 500.
58. Munakata K, Koi M, Kitajima T, Tseng-Rogenski S, Uemura M, Matsuno H, et al. Inflammation-associated microsatellite alterations caused by MSH3 dysfunction are prevalent in ulcerative colitis and increase with neoplastic advancement. Clinical and Translational Gastroenterology. 2019 Dec;10(12).
59. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Research. 1998 Nov 15;58(22):5248-5257.
60. Watson MM, Berg M, Søreide K. Prevalence and implications of elevated microsatellite alterations at selected tetranucleotides in cancer. British Journal of Cancer. 2014 Aug;111(5):823-7.
61. Kim CH, Wu W, Wysoczynski M, Abdel-Latif A, Sunkara M, Morris A, et al. Conditioning for hematopoietic transplantation activates the complement cascade and induces a proteolytic environment in bone marrow: a novel role for bioactive lipids and soluble C5b-C9 as homing factors. Leukemia. 2012;26(1):106-16.
62. Hile SE, Shabashev S, Eckert KA. Tumor-specific microsatellite instability: do distinct mechanisms underlie the MSI-L and EMAST phenotypes?. Mutation Research/ Fundamental and Molecular Mechanisms of Mutagenesis. 2013 Mar 1;743:67-77.
63. Boland CR, Sinicrope FA, Brenner DE, Carethers JM. Colorectal cancer prevention and treatment. Gastroenterology. 2000 Feb 1;118(2):S115-28.
64. Carethers JM. Systemic treatment of advanced colorectal cancer: tailoring therapy to the tumor. Therapeutic Advances in Gastroenterology. 2008 Jul;1(1):33-42.
65. Tajima A, Hess MT, Cabrera BL, Kolodner RD, Carethers JM. The mismatch repair complex hMutSrecognizes 5-fluoruracil-modified DNA: implications for chemosensitivity and resistance. Gastroenterology 2004;127:1678-1684.
66. Tajima A, Iwaizumi M, Tseng-Rogenski S, Cabrera BL, Carethers JM. Both hMutSa and hMutSß complexes participate in 5-fluoruracil cytotoxicity. PLoS One 2011;6:e28117.
67. Iwaizumi M, Tseng-Rogenski S, Carethers JM. DNA mismatch repair proficiency executing 5-fluorouracil cytotoxicity in colorectal cancer cells. Cancer Biology & Therapy. 2011 Oct 15;12(8):756-64.
68. Carethers JM, Smith EJ, Behling CA, Nguyen L, Tajima A, Doctolero RT, et al. Use of 5-fluorouracil and survival in patients with microsatellite-unstable colorectal cancer. Gastroenterology. 2004 Feb 1;126(2):394-401.
69. Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, et al. Tumor microsatelliteinstability status as a predictor of benefit from fluorouracilbased adjuvant chemotherapy for colon cancer. New England Journal of Medicine. 2003 Jul 17;349(3):247-57.
70. Hamaya Y, Guarinos C, Tseng-Rogenski SS, Iwaizumi M, Das R, Jover R, et al. Efficacy of adjuvant 5-fluorouracil therapy for patients with EMAST-positive stage II/III colorectal cancer. PloS one. 2015 May 21;10(5):e0127591.
71. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017 Jul 28;357(6349):409-13.
72. Koi M, Carethers JM. The colorectal cancer immune microenvironment and approach to immunotherapies. Future Oncology. 2017 Aug;13(18):1633-47.