Abstract
Stimulating effector T-cells (Teffs) without inducing regulatory T-cells (Tregs) has been the primary goal of IL-2-based therapies for cancer. Recently, modified IL-2 designed for differential T-cell expansion for the treatment of cancer has failed in the clinic. We propose that treatments based on exogenous administrations of modified IL-2 are inherently undermined by a negative feedback loop, caused by IL-2 secreted endogenously from activated effector T-cells. This endogenous IL-2 secretion subsequentially induces Treg expansion and inhibits the immune response that is essential for cancer clearance. Here, we demonstrate that treatments utilizing exogenous modified IL-2 indeed induce Treg expansion. To circumvent this negative feedback, we computationally designed a novel monoclonal humanized antibody (AU-007) that binds human IL-2 with pM affinity at a predefined epitope and completely blocks IL-2 binding to CD25 that is highly expressed on Tregs, without hindering IL-2 binding to CD122/CD132 dimer receptor expressed over effector cells. This epitope-specific, high-affinity antibody controls endogenous IL-2 and prevents it from expanding Tregs while allowing it to expand Teffs. We show that controlling endogenous IL-2 using AU-007 abrogates the negative feedback loop and replaces it with a positive feedback loop that enhances the expansion of NK cells and Teffs, an effect considered favorable for cancer immunotherapy.
Keywords
IL-2, Modified IL-2, Cancer immunotherapy, Anti-tumor therapy, Targeting immunotherapy, Immune cells response, Regulatory T cells (Tregs), Effectors T cells (Teffs), Natural Killer cells (NK), IL-2-induced IL-2 release feedback loop
Introduction
Interleukin-2 (IL-2) is a key mediator in the expansion and activation of T-cells and natural killer cells (NKs). IL-2 plays a major role in the secondary signals required for T-cell activation. It binds two forms of the IL-2 receptor: a high-affinity trimeric receptor composed of CD25, CD122, and CD132, and a low-affinity dimeric receptor composed of CD122/CD132 [1]. T effector cells (Teffs) and NKs can signal via IL-2 binding to the dimeric receptor, leading to their expansion and activation. Regulatory T-cells (Tregs) rely on the high-affinity trimeric receptor complex to enhance their functions, which include immunosuppressive cytokine release, mitotic expansion, and sequestering IL-2 away from binding to memory and naive T-cells [2]. Activated Teffs secrete IL-2 [3] and transiently upregulate CD25 expression [4]. The binding of IL-2 to these activated Teffs induces restimulation-induced cell death (RICD) [5,6]. IL-2 is produced by activated CD4+ T helper cells and activated CD8+ Teffs [7,8]. Thus, effector cells that are expanded by IL-2 secrete more IL-2 upon activation. Autocrine and paracrine secretion of IL-2 from activated T-cells also reduces immune activation by increasing RICD of activated Teffs and by enhancing the expansion and fitness of Tregs [9]. As such, immune activation by IL-2 is under tight negative feedback regulation through IL-2-induced IL-2 release.
Overall, IL-2 has a dual effect in regulating the immune response. During homeostasis, low levels of IL-2 are available to bind and stimulate the high-affinity trimeric receptor expressed over Tregs [1]. IL-2 is also the predominant cytokine that is produced in high concentrations by TCR-activated and IL-12-stimulated Th1 cells during a primary response [10]. These increasingly high levels of IL-2 interacting with the dimeric receptor expressed on naïve and memory precursor (MP) T cells are required for the proliferation and differentiation of these cells into effector cytotoxic cells, and these T cells also secrete IL-2 when activated [11]. In addition, IL-2 is known to have a role in shutting down the immune response and restoring homeostasis [12-14]. TCR-activated and IL-2-stimulated T cells transiently upregulate CD25 expression [15], and those short-lived CD25+ cytotoxic T cells are prone to apoptosis in response to a second stimulation of IL-2 [10]. IL-2 mediates reactivation-induced T-cells death (AICD) [16,17] also known as RICD, while CD25 blockade protects T cells from dying [18]. Tregs, do not secrete IL-2, stably express high levels of CD25, and are solely dependent on IL-2 for survival [19]. Thus, IL-2 modulates the immune response via a negative feedback loop mechanism. T-cell TCR activation leads to IL-2 secretion to support immune stimulation, while a second response to IL-2 on these TCR-activated and IL-2 stimulated terminally differentiated T cells leads to their death, yet maintains the viability and functionality of Tregs to support immune suppression. The IL-2 proinflammatory effect is crucial for cancer clearance due to the cytotoxic activity of the effector cells, while the IL-2 immune inhibitory effect undermines it.
High-dose IL-2 (HD IL-2) is approved for the treatment of melanoma and renal cell carcinoma, but its therapeutic value is limited due to limited efficacy, severe toxicity, and short in vivo half-life [1]. The major challenge in the development of IL-2 as a therapeutic antitumor agent is that IL-2 can act on both effector cells that express the dimeric receptor and Tregs that express the trimeric receptor [20]. Cancer patients given a high dose of IL-2 showed limited efficacy due to increased Treg levels [21]. Stimulating Teffs and NKs without inducing Tregs has been the main challenge in developing IL-2-based therapies for oncology. Modifying IL-2 to have a biased selectivity for cells that express IL-2 dimeric receptor and prevent its interactions with Tregs aims to solve traditional IL-2 therapy’s limited efficacy. In recent years, several investigational therapeutics that are based on modified IL-2 have been developed with the goal of eliminating the inhibitory function of IL-2 while maintaining its activating effect. These approaches utilize exogenously administered modified IL-2 that preferentially activate Teffs relative to Tregs due to modifications that limit IL-2’s ability to bind the CD25 subunit [22,23]. However, a primary source of endogenous IL-2 is activated Teffs [24-27]. Modified IL-2 with a bias selectivity to the dimeric receptor, indeed leads to the proliferation of effector T-cells but has no effect on the action of the native endogenous IL-2 that is secreted from the same activated T cells that it acts on. Thus, this newly endogenous IL-2 is free to shut down the immune response and undermine the modified IL-2 effect. We hypothesized that exogenous engineered IL-2 expands cells that secrete endogenous IL-2, creating negative feedback by expanding Tregs. Specifically, modified cytokine approaches lack the ability to control the autocrine/paracrine-secreted IL-2 and their efficacy will be limited by the expansion of Tregs and enhanced RICD triggered by endogenously released, unmodified, IL-2. We further hypothesized that controlling endogenously secreted IL-2 will prevent this autoinhibitory feedback loop and replace it with a positive feedback loop that will improve efficacy. To test this hypothesis, we compared the activity and effect of three compounds: non-modified IL-2, a non-alpha-IL-2 (naIL-2, IL-2 conjugated to CD25), and a high-affinity anti-IL-2 monoclonal antibody (AU-007) that completely block IL-2 interaction with CD25 while sparing its interaction with the dimeric CD122/CD132 receptor (Figure S1). We found that treatment with exogenous IL-2 or naIL-2 resulted in a downstream expansion of Tregs (Figure 2) while controlling endogenous IL-2 using an epitope-specific blocking Ab (AU-007) resolved IL-2 immune inhibitory negative feedback loop and replaced it with a positive feedback loop that is preferred for cancer immunotherapy (Figures 1-2).
Results
Generation of IL-2-based compounds that inhibits IL-2 binding to CD25 and allow IL-2 signaling only through the dimer receptor
To demonstrate the effect of the IL-2 negative feedback loop, we developed two compounds: i) a monoclonal antibody (AU-007) that captures endogenously secreted IL-2 and blocks its ability to bind and activate IL-2 trimeric receptor expressed over regulatory T-cells while sparing IL-2 interactions with the dimeric receptor expressed over effector cells. ii) and a non-alpha-IL-2 (naIL-2) cytokine that can only interact with the dimeric receptor yet has no effect over endogenous IL-2 that is natively secreted from activated T-cells. AU-007 was computationally designed to bind IL-2 at the CD25 binding site. The epitope differentiates the binding of the dimer on effector cells from the epitope that mediates the binding on the trimer expressed on Tregs was inferred from the crystal structure (PDB code 2B5I). We focused on the surface of IL-2 that is buried under CD25 and avoided surface residues that contact the dimer subunits. Using surface plasmon resonance, we verified that AU-007 binds hIL-2 with a desired pM affinity required for capturing low levels of IL-2 that allows full competition with the high-affinity trimeric receptor (Figure S1.A). Next, we showed that both the naIL-2 and AU-007 inhibited binding to CD25 but not to CD122 (Figure S1.B-D). Further, we showed that both AU-007 and the naIL-2 cytokine preserve IL-2 functional signaling through the IL-2-dimeric receptor. To demonstrate this, we used a reporter cell line that stably expresses the human IL-2 dimer receptor (CD122/CD132) and showed that neither AU-007 nor naIL-2 interfered with IL-2/IL-2-dimer receptor signaling (Figure S2).
AU-007 binds to endogenous IL-2 and breaks the negative feedback loop in human PBMCs
In order to break the autoinhibitory feedback loop of IL-2 in inflamed systems, it is not sufficient to bind IL-2 with tight affinity and block its interaction with CD25. An additional and challenging requirement is that the antibody must acquire the ability to capture and control endogenous IL-2 that is being constitutively secreted by activated T cells and consumed by CD25+ Tregs or activated CD25+ Teffs. To validate that AU-007 captures endogenously secreted hIL-2 in vivo, immunodeficient NOG-EXL mice were engrafted with hPBMCs. Ten days post engraftment, mice were treated once with AU-007 or with an isotype control antibody. Eight days post-treatment hearts were terminally bled for serum collection and spleens were harvested for immune cell analysis using flow cytometry (Figure S3.A). AU-007 single-dose administration led to the accumulation of endogenously secreted hIL-2 and inhibited Tregs. Significant high levels of AU-007 bound to endogenous IL-2 were detected from mouse serums (Figure S3.B). CD8+ T-cells and NKs were elevated compared to the isotype control-treated mice even eight days post-treatment (Figures S3.C-G). To further test whether AU-007 efficiently captures endogenous IL-2 in vitro, we treated hPBMCs with AU-007. Total hPBMCs were treated once with AU-007 or with an isotype control antibody, without adding exogenous IL-2. Cells were monitored daily for 7 days by flow cytometry. While the control antibody didn’t affect Tregs, AU-007 completely inhibited the expansion of Tregs (Figure 1A) and significantly increased Teff:Treg ratio (Figure 1B). Next, we activated hPBMCs with anti-CD3/anti-CD28 and treated the activated culture with AU-007. Under these T-cell activating conditions, AU-007 selectively inhibited Tregs without affecting Teffs or NKs. Furthermore, AU-007 downregulated the suppressive markers of CD4+Treg (Figures 1D-1E, 1J and 1K). Since no exogenous hIL-2 was added, this indicates that AU-007 captures endogenous IL-2 and prevents the negative feedback loop initiated by the IL-2 secreted from activated T-cells that leads to the expansion of Tregs.
AU-007 can capture and redirect endogenous IL-2 to break the auto-inhibitory loop in hPBMCs while HD IL-2 or naIL-2 cannot
Finally, the negative feedback loop caused by IL-2 or naIL-2 was compared to the positive feedback loop caused by AU-007. Total hPBMCs were incubated in vitro for 7 days after a single treatment with either 1 nM of naIL-2 alone or 1 nM of IL-2 combined with AU-007 in two different ratios 1:1000 (1uM of AU-007) or 1:10,000 (10 uM of AU-007). Immune cell subpopulations were analyzed daily by flow cytometry. Figure 2 shows that both AU-007 and naIL-2 expanded NKs and Teffs. However, the addition of naIL-2 led to the expansion of Tregs even several days after its administration, whereas the addition of AU-007 did not. These data demonstrate that AU-007 redirects both exogenous IL-2 and endogenous paracrine secreted IL-2 in a way that inhibits Tregs and the expression of Treg suppressive markers while increasing Teffs:Tregs ratio and expanding NKs (Figures 2A-2E). To maintain homeostasis upon activation of T-cells, paracrine IL-2 induces Tregs expansion. However, both autocrine and paracrine IL-2 induce RICD on activated CD25+ T-cells. To test if AU-007 protects cells from RICD, we examined the overall viability of activated hPBMCs in the presence or absence of AU-007. hPBMCs were stimulated once with anti-CD3/anti-CD28 antibodies with or without 10 uM of AU-007. Three days post-stimulation, samples were treated with IL-2 (1nM) to induce IL-2-dependent RICD. Lymphocyte viability was monitored daily for 14 days using flow cytometry. Stimulated lymphocytes showed a 50% reduction in viability after a single dose of IL-2. The addition of AU-007 caused a ~10% increase in viability, further supporting the ability of AU-007 to prevent the stimulus-induced negative feedback caused by endogenous IL-2 release (Figures 2F-2H). Taken together, these results show that activation-induced release of IL-2 produces an immunosuppressive effect by expanding Tregs and triggering RICD. This activation-induced release is triggered by IL-2 itself, producing a self-delimiting negative feedback loop, which interferes with the stimulatory effect of exogenously administered IL-2 or naIL-2. AU-007, a computationally designed antibody that prevents IL-2 binding to CD25, redirects autocrine/paracrine secreted IL-2, thereby converting the negative feedback loop into a positive feedback loop that prolongs immune stimulation. The IL-2 autoinhibitory feedback loop described here may explain the limited efficacy of modified IL-2 therapies observed in clinical development. Moreover, our findings indicate that therapeutic approaches targeting the IL-2 autoinhibitory feedback loop may hold promise for cancer immunotherapy. AU-007 is currently being evaluated in a Phase 1/2 clinical trial in several types of tumors (ClinicalTrail.gov identifier: NCT05267626).
Materials & Methods
Non-alpha-IL-2 (naIL-2) production
Human IL-2 with an 8xHis C-terminal tag conjugated with a linker (G4STRG4STG4SG4SS) to human CD25 extracellular domain at the IL-2 N-terminus was expressed in Expi293 cells (ThermoFisher, A14527) using pSF-CMV expression vector according to the manufacturer protocol. In brief, 1 ug/ml DNA was transfected to cell culture at a density of 3×106 (viability of 97%) using an ExpiFectamin293 kit containing transfection reagent and enhancers (ThermoFisher, A14524). After transfection, cells were incubated at 37oC with 80% humidity and 8%CO2. Enhancers were added to the transfected cell culture 21 hours post-transfection. 7 days post-transfection, cells were harvested, and the supernatant was collected and dialyzed overnight to PBS supplemented with 25 mM imidazole and 200 mM NaCl (buffer A). The following day, the supernatant was loaded on 5 ml HisTrap (Cytiva, 17524802) at a flow rate of 2.5 ml/min. Following buffer A wash, elution was done in steps 20, 40, and 60% buffer B containing PBS, 200 mM NaCl, and 0.5 M imidazole at a flow rate of 4.5 ml/min. After SDS-PAGE analysis, fractions were pooled and dialyzed overnight to PBS. The non-alpha-IL-2 protein was flash-frozen in liquid N2 at 1 mg/ml.
SPR Kinetics for determination of AU-007 affinity to IL-2
Kinetic measurements of AU-007 for human IL-2 were done using Biacore T200 (GE Healthcare, USA) on a CM5 chip (BR10005-30, Cytiva). The chip was crosslinked with a human antibody capture kit (BR100839, GE Healthcare, USA) according to the manufacturer’s protocol to target 3000-4000 RU. Kinetics determination was performed using a multi-cycle strategy, AU-007 was captured on the coated chip to reach ~300 RU. Human IL-2 (RKP60568, Reprokine) was injected from 10 nM to 0.003 nM in two-fold dilutions. Between each cycle, all channels underwent regeneration using 3 M MgCl2. Binding kinetics were determined by the 1:1 Binding model using the Biacore T200 evaluation software, version 3.1.
SPR for determination of AU-007 binding epitope on IL-2. AU-007 was captured on an anti-human Fc (BR100839, Cytiva) coated CM5 chip (BR10005-30, Cytiva) to reach ~300 RU. 50 nM human IL-2 (RKP60568, Reprokine) was injected over 60 seconds to saturate the antibody, and then either 1uM CD25 (RKP01589, Reprokine) or 1uM CD122 (RKP14784, Reprokine) was injected for 60 seconds. AU-007 binding epitope was characterized based on the ability of the IL-2 receptor subunits to bind IL-2 in the presence of AU-007.
SPR for determination of non-alpha-IL-2 available epitopes
To test whether the CD25 epitope is blocked in the non-alpha-IL-2 format, a biotinylated human CD25 (AVI10305-050, R&D systems) was captured on a streptavidin-coated chip (BR100531, Cytiva) and 200 nM of human IL-2 (RKP60568, Reprokine) or non-alpha-IL-2 (IL-2 conjugated to CD25 subunit) were injected for 60 seconds. To verify that the ability of the non-alpha-IL-2 to bind CD122 is preserved, an Fc-tagged CD122 (extracellular domain) (RKP14784F, Reprokine) was captured on an anti-human Fc-coated (BR100839, Cytiva) CM5 chip (BR10005-30, Cytiva) to reach ~400 RU and 1 uM of human IL-2 (RKP60568, Reprokine) or the non-alpha-IL-2 were injected for 90 seconds. In both assays, CD25 and CD122 available epitopes were characterized based on the ability of the IL-2 receptor subunits to bind IL-2 or the non-alpha-IL-2.
Naïve Human PBMCs expansion assay
Human PBMCs were thawed into complete hPBMC media (RPMI, supplemented with 10% FBS, 1% pen/strep, 1% sodium pyruvate, 1% NEAA, 1% Glutamax, 0.1% 2-mercaptoethanol – 0.05mM), and rested for 4-5 h prior to assay initiation. After resting, cells were cultured in 24 well plates (1×106 cells/1ml in each well) with 1 uM of AU-007 or with 1 uM of an isotype control antibody without supplementation of exogenous hIL-2 (Figures 1A-1E). Alternatively, cells were incubated with 1 nM of non-alpha-IL-2 or 1nM native hIL-2 in the presence of 1 uM or 10 uM of AU-007 or an isotype control antibody (Figures 2A-2E). Cells were monitored for 7 days, and immune cell subpopulation analysis was done daily using flow cytometry. At each time point, cells were stained with fixable viability dye according to manufacturer instructions, followed by staining of cell surface markers (CD3, CD4, CD8, CD25, CD56, CD127), fixation /permeabilization (Miltenyi, cat# 130-093-142) followed with staining of the intracellular marker, FoxP3. Tregs were defined as CD3+CD4+CD25+CD127-FoxP3+, CD8 Teffs were defined as CD3+CD8+FoxP3-, NK cells were defined as CD3-CD56+, NKTs were defined as CD3+CD8+CD56+. Gating was defined based on fluorescence minus one control (FMO); all gating originated from lymphocytes' live cells. The full antibodies list that was used for the detection of immune cell markers is detailed in Table S1.
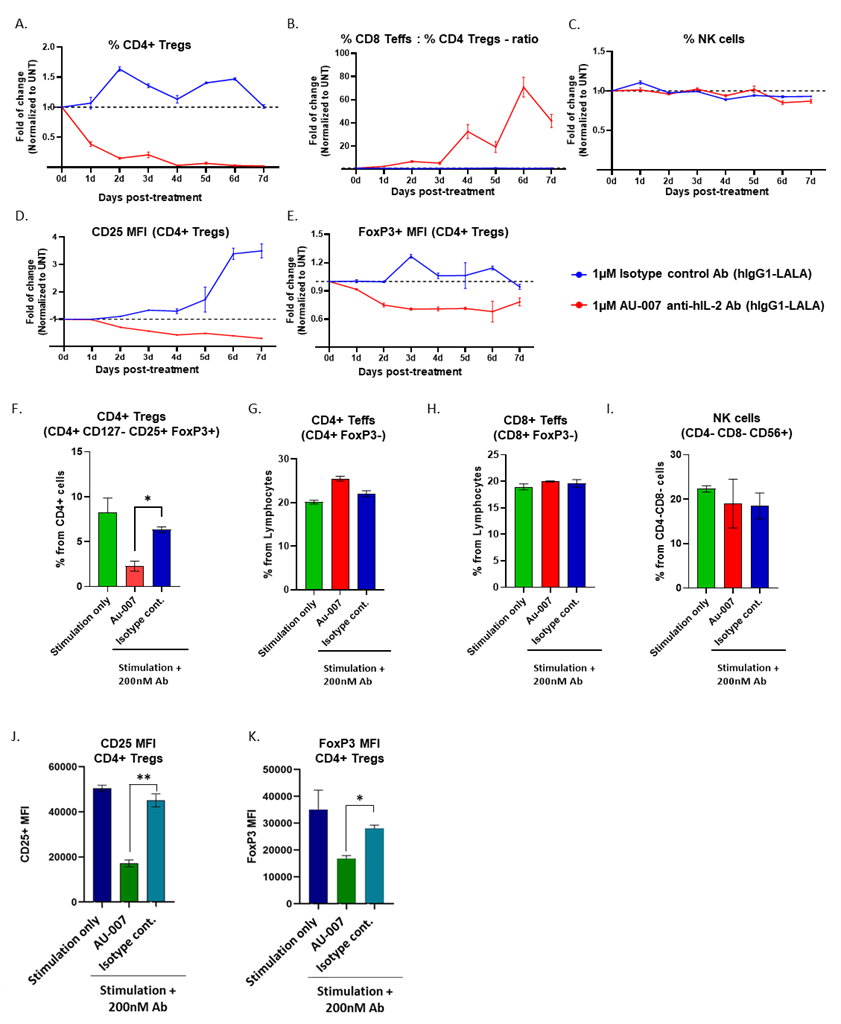
Figure 1. AU-007 binds to endogenous IL-2 and breaks the negative feedback loop in human PBMCs. A-E: naive hPBMCs were treated once at day 0 with either 1uM AU-007 (red) or with an isotype control antibody (blue). No exogenous IL-2 was added. The culture was monitored for 7 days, and immune cell subpopulations were analyzed daily by flow cytometry. Values were normalized to untreated samples (UNT) at each day. AU-007 completely inhibits Tregs expansion (A) and significantly increases Teffs:Tregs ratio (B), without hindering NKs (C). AU-007 downregulates the suppressive markers of CD4+Treg from panel A, as defined by a significant reduction in mean fluorescence intensity (MFI) of CD25 and FoxP3 (D-E). F-K: Total hPBMCs were stimulated for 24h with anti-CD3/anti-CD28 (stimulation only, green) or stimulated with anti-CD3/anti-CD28 in the presence of 200nM of AU-007 mAb (red) or with 200nM of isotype control mAb (blue). No exogenous IL-2 was added. Immune cells subpopulations were analyzed by flow cytometry. AU-007 inhibits Tregs without hindering effector cells and NKs (F-I). AU-007 downregulates the suppressive markers of CD4+Treg from panel G, as defined by a significant reduction in MFI of CD25 and FoxP3 (J-K).
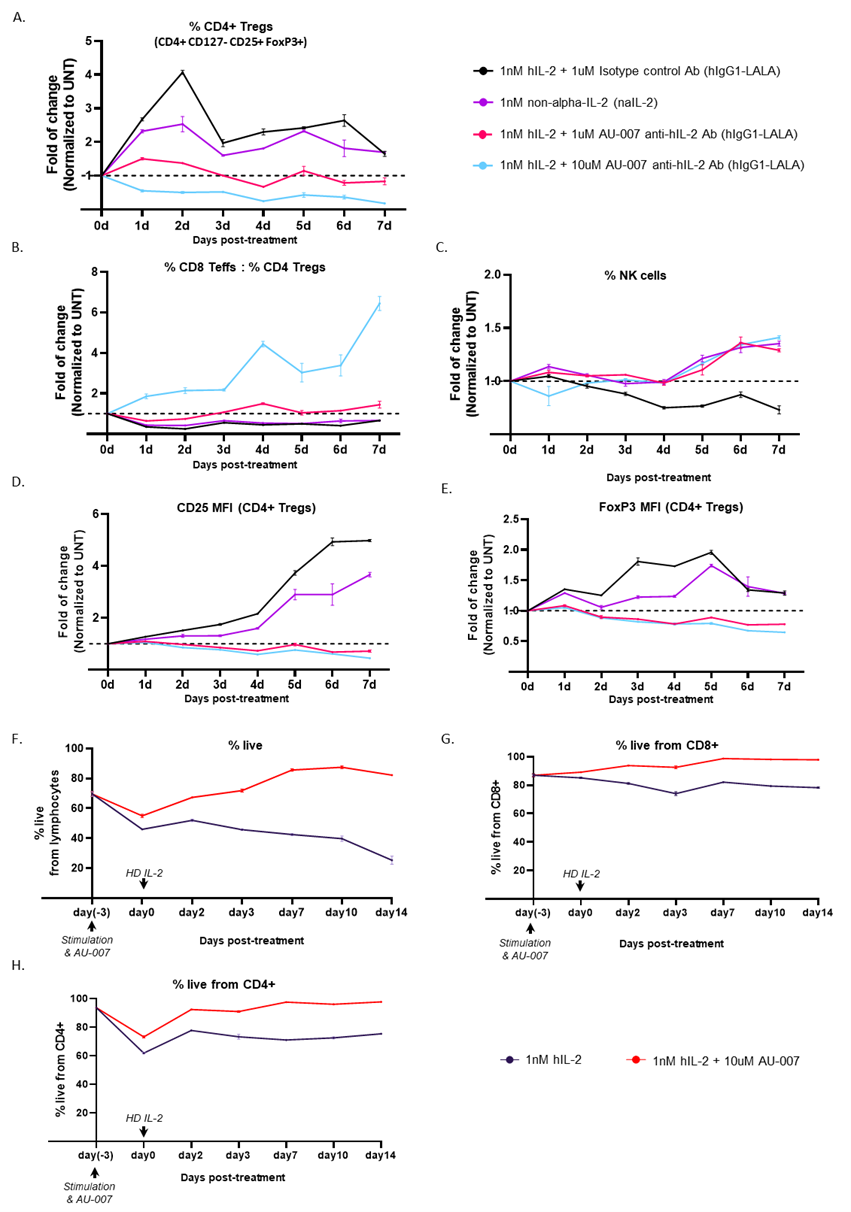
Figure 2. AU-007 can capture and redirect endogenous IL-2 to break the auto-inhibitory loop in hPBMCs while HD IL-2 or naIL-2 cannot. AU-007 promotes the expansion of NKs and CD8 T-cells while completely inhibiting the expansion of regulatory T-cells. A-E: naive hPBMCs were treated once on day 0 with 1nM of naIL-2 (purple) or with HD IL-2 (1nM) combined with 1uM of isotype control Ab (black) or with either 1uM AU-007 (red) or 10uM AU-007 (turquoise). The culture was monitored for 7 days, and immune cell subpopulations were analyzed daily by flow cytometry. Values were normalized to untreated samples (UNT) at each day. While naIL-2 expands NKs similarly to AU-007 it fails to inhibit Tregs expansion, while AU-007 completely inhibits Tregs expansion in culture (A) and significantly increases the Teffs:Tregs ratio (B), without hindering NKs (C). AU-007 downregulates the suppressive markers of CD4+Treg from panel A, as defined by a significant reduction in MFI of CD25 and FoxP3 (D-E). F-H: AU-007 rescues activated lymphocyte viability decreased by treatment with HD IL-2. hPBMCs culture was stimulated once with anti-CD3/anti-CD28 Abs with or without 10uM of AU-007. 3 days post-stimulation all samples were given HD IL-2 (1nM) and were monitored daily for cell viability using flow cytometry.
Activated Human PBMCs expansion assay
Human PBMCs were thawed into complete PBMC media and rested overnight prior to assay initiation. After resting, cells were activated with anti-CD3/anti-CD28 Abs (Stemcell, cat#10991) and cultured in 24 well plates (1×106 cells/1 ml in each well) with 200nM of either AU-007 or an isotype control antibody for 24 h. After incubation, cells were stained with fixable viability dye according to manufacturer instructions, followed by extracellular markers staining (CD4, CD8, CD25, CD56, CD127), fixation/permeabilization (Miltenyi, cat# 130-093-142) and staining of the intracellular marker FoxP3 for the assessment of different cell subsets expanded after treatment. Tregs were defined as CD4+CD25+CD127-FoxP3+, CD8 Teffs were defined as CD8+FoxP3-, NK cells were defined as CD8-CD4-CD56+, NKTs were defined as CD8+CD56+. Gating was defined based on fluorescence minus one control (FMO); all gating originated from lymphocytes' live cells. The full antibodies list that was used for the detection of immune cell markers is detailed in Table S2.
Lymphocyte viability assay
Human PBMCs were thawed into complete PBMC media and rested for 4-5 h prior to assay initiation. After resting, cells were activated with anti-CD3/anti-CD28 Abs (Stemcell, cat#10991) and cultured in 24 well plates (1×106 cells/1 ml in each well) with or without 10 uM of AU-007. 3 days later cultures were supplemented with 1 nM hIL-2 and monitored by flow cytometry at 7- and 14-days post-treatment. At each time point, cells were stained with fixable viability dye according to manufacturer instructions, followed by extracellular markers staining (CD4, CD8) and viability die.
HEK dimer pSTAT5 activation
HEK cells expressing the dimeric form of the IL-2 receptor (hkb-IL-2, Invivo-Gen) were grown in complete media (DMEM (high glucose,) + 1% L-glutamine) until harvesting on the day of the experiment. Upon cell count, 50,000 cells/100 ul were seeded per well in a 96-well plate. Cells were treated with serial dilutions of non-alpha-IL-2 (100nM-1.28 pm) or with serial dilutions of hIL-2 (100 nM-1.28 pM) combined with 200 nM of either AU-007 or an antibody that inhibits IL-2 binding to the dimeric receptor (dimer inhibitor antibody) or with an isotype control antibody. Treatment was done in a final volume of 200 ul/well. The cells were then transferred to 37°C for overnight incubation. After 24 h, 20 ul of cell supernatant was transferred to a new plate and 180 ul of Quanti-Blue (Invivo-Gen, Cat#: rep-qbs) working solution was added to the wells. The plate was incubated with Quanti-Blue for 1 h and analyzed using a plate reader at 620-655 nm.
Mice study
Animal studies were held at Crown Bioscience, Inc. (San Diego). Mice were housed under specific pathogen-free conditions as per the national animal testing regulations. Animal welfare for this study complies with the U.S. Department of Agriculture’s Animal Welfare Act in strict accordance with applicable Crown Bioscience, Inc. 10 days prior to dosing, 6-9 weeks of age female NOG-EXL non-humanized mice (Jackson Laboratory) were inoculated with 10 million PBMCs (isolated from 3 different donors) per mouse via i.v. injection. Before dosing, all animals were randomly allocated to the different study groups, each group contained 9 mice, 3 mice for each hPBMCs’ donor. Randomization was performed in the Study Log software two days prior to dosing [28]. The average body weight (grams) for each group ± SD at randomization was as follows: Group 1 - Isotype control 17.37 ± 1.09, Group 2 AU-007 17.47 ± 0.94. Animals were dosed once with 20 mg/Kg (5 ml/Kg) via i.p. Eight days post-treatment animals were harvested, hearts were terminally bled for serum collections, and spleens were collected for immune cell analysis.
Ab/hIL-2 complex detection from mice serums
Ab/hIL-2 complex was detected from mice serums using ELISA. High binding 96 well plate was coated with 200 ng/well polyclonal goat anti-hIL-2 and incubated overnight at 4oC (R&D Bioscience, Kit catalog: #SEL202, part: #840606 Lot: QQ0819051). The plate was then washed 3 times with 300 ul PBS-T (0.05% Tween) followed by blocking using 1.5% Milk diluted in PBS-T at 300 ul/well and incubated for 1 h at R.T. Plate was washed 3 times with 300 ul PBS-T (0.05% Tween) and incubated for 1 h at room temperature with serums samples or with standard curve samples. Pre-incubation, serums were diluted 1:2 according to the MRD and compared to a standard curve of AU007 mixed with hIL-2 at 2:1 ratio respectively in serum diluted at room temp according to the MRD. The plate was washed 3 times with 300 ul PBS-T (0.05% Tween) and incubated for 30 minutes at room temperature with goat anti-human Fc-HRP diluted 1:20000 in PBS (Jackson ImmunoResearch 109-035-008). The plate was washed again 3 times with 300 ul PBS-T (0.05% Tween). Chromogenic substrate (TMB) was added for each well followed by a stop solution. The plate was analyzed using a plate reader at 450 nm.
Discussion
IL-2 was the first successful cancer immunotherapy. As early as 1984 it had been observed that, while extremely toxic, high doses of IL-2 can cure advanced metastatic cancer in some patients [29]. Ever since, harnessing IL-2 to treat cancer has been a promising and challenging goal [30]. Recombinant hIL-2 (Proleukin, aldesleukin) was approved as a cancer immunotherapeutic in the 1990s. It showed efficacy in some individuals that tolerated the high toxicity of the high doses required to induce antitumor effects [29,30]. The basis behind the immunomodulatory biology of IL-2 is dependent on differential concentrations of the cytokine during homeostasis and inflammation. At low concentrations, the cytokine binds a trimeric IL-2 receptor on regulatory T cells, whereas at high concentrations, IL-2 will also engage the lower-affinity dimeric receptor found on effector T cells and natural killer cells [31]. The main source of endogenous IL-2 is activated T cells [10]. These cells secrete IL-2 while also transiently upregulating the CD25 subunit to express the high-affinity trimeric receptor [15,16]. The secreted IL-2 is consumed by Tregs as well as by the activated effector cells. The effect of the consumed IL-2 is radically different between these T-cell populations. IL-2 consumption by Tregs and naïve Teffs leads to their expansion, while its consumption by activated CD25+ Teffs is believed to induce apoptosis [32-34]. In both cases, IL-2 helps restore homeostasis by downregulating the immune attack (Figure 3A). Multiple attempts have been made to modify IL-2 in a way that will bias its selectivity toward the expansion of Teffs and away from Tregs. While these therapeutic approaches had shown great promise at pre-clinical stages, their clinical results have been disappointing [35,36], (Table 1). Here we show that treatments involving an external source of IL-2 or a naIL-2 with biased selectivity to effector cells, fail to handle the negative feedback loop of endogenous IL-2 that inherently drives the immune system toward immune downregulation and homeostasis. Although naIL-2 cannot interact with CD25 (Figure S1.C), binds CD122 (Figure S1.D), stimulates the dimeric receptor properly (Figure S2), and expands NK cells (Figure 2C), it fails to prevent Tregs expansion (Figure 2A). The fact that CD25+ Tregs, that rely solely on IL-2 to survive, double their percentage and remain viable even 7 days post-treatment, it clearly shows that the naIL-2 activity fails to control the natively expressed IL-2 that is free to bind CD25+ Tregs (Figure 3B). We demonstrate that AU-007, a monoclonal antibody that captures and controls the activity of endogenous secreted IL-2 produced by activated T cells, can break this autoinhibitory feedback loop of IL-2 and drive its activity towards immune stimulation. AU-007 captures and redirects endogenous IL-2, allowing it to expand Teffs cells and NK cells while completely preventing it from binding CD25+ cells. As demonstrated in Figure 1 and Figure S3, activated cells that were treated once with AU-007 inhibit Tregs and maintain Teffs and NK cells even without the addition of IL-2 from an external source. These results demonstrate that AU-007 captured and controlled endogenously secreted IL-2, preventing it from stimulating Tregs, while allowing it to effectively stimulate Teffs and NK cells. Moreover, this long-term effect was also observed when cells were treated once with AU-007 followed by a single high dose of IL-2 (Figures 2A-2E). Additionally, Figure 2F-2H shows that AU-007 rescues the activated and IL-2-stimulated lymphocytes from cell death. The inhibition of Tregs, the expansion of effector cells, and the prevention of activated effector cell death all together break the autoinhibitory activity of IL-2. This allows for the expansion of the immune stimulatory stage that is considered favorable for cancer immune therapy [37- 40], (Figure 3C). Dozens of engineered IL-2 therapies are still being investigated in clinical trials. Based on the results presented here we suspect that therapeutic approaches that are based on external administrations of IL-2 will face the same limitation driven by the uncontrolled endogenously secreted IL-2. AU-007, which is also in clinical development, may remedy this by redirecting endogenous IL-2 selectively to Teffs. As shown here, AU-007, eliminates the negative feedback loop that undermines the effect of modified exogenous IL-2, and may even replace it with a positive feedback loop that reinforces the expansion of Teffs by IL-2. This control of the activity of endogenously secreted IL-2 has the potential to modulate the immune response in a more precise and targeted manner to improve treatment outcomes. Since AU-007 promotes the expansion of Teffs and NK cells, we expect it to show efficacy as a monotherapy and, we also hypothesize that combining it with either immune checkpoints inhibitors (ICI) like anti-PD-1 or with ADCC agents (antibody-dependent cell-mediated cytotoxicity) like anti-Her2 or anti-PD-L1 will result in increased efficacy. Additionally, AU-007 potentially also be used in combination with T-cells and NK cells therapies, such as; adoptive transfer, CAR-Ts, and engineered NK cells to improve the expansion and survival of these cells. In conclusion, the finding of this study proposes a possible explanation for the recent setbacks of modified IL-2 compounds in immune-oncology. We suggest that improving IL-2-based therapy must include a way to control the activity of endogenous IL-2. Epitope specific IL-2 antibodies like AU-007 are great examples of such a mechanism.
|
Drug |
Company |
Description |
Phase |
|
?AU-007 NCT05267626 |
Aulos Bioscience |
Anti-IL-2 mAb (controlling endogenous IL-2) |
1 / 2 (ongoing) |
|
*Bempegaldesleukin NCT04410445 |
Nektar Therapeutics |
PEGylated IL-2 mutein |
3 (Discontinued) |
|
*Nemvaleukin alfa NCT04830124 |
Alkermes |
IL-2–CD25 fusion protein |
3 (ongoing) |
|
*SAR444245 NCT05535023 |
Sanofi |
PEGylated IL-2 mutein |
2 (Discontinued) |
|
*ANV419 NCT04855929 |
Anaveon |
IL-2–anti-IL-2 fusion protein |
1 / 2 (ongoing) |
|
*MDNA11 NCT05086692 |
Medicenna |
Albuminated IL-2 mutein |
1 / 2 (ongoing) |
|
*TransCon NCT05081609 |
IL-2 Ascendis Pharma |
IL-2 Ascendis Pharma |
1 / 2 (ongoing) |
|
*BNT151 NCT04455620 |
BioNTech |
mRNA-encoded IL-2 mutein |
1 / 2 (ongoing) |
|
*XTX-202 NCT05052268 |
Xilio |
Masked IL-2 mutein |
1 / 2 (ongoing) |
|
*NL-201 NCT04659629 |
NeoleukinTherapeutics |
CD25 independent IL-2/IL-15 agonist |
1 (Discontinued) |
|
*STK-012 NCT05098132 |
Synthekine |
α/β-selective IL-2 mutein |
1 (ongoing) |
|
*CUE-101 NCT03978689 |
Cue Biopharma |
E7-pHLA-IL-2-Fc fusion protein |
1 (ongoing) |
|
*RG6279 NCT04303858 |
Roche |
IL-2–anti-PD-1 fusion protein |
1 (ongoing) |
|
*MK-1484 NCT05382325 |
Merck |
IL-2–anti-PD-1 fusion protein |
1 (ongoing) |
|
*SHR-1916 NCT04842630 |
Jiangsu Hengrui |
PEGylated IL-2 mutein |
1 (ongoing) |
|
*Therapies that are based on the administration of exogenous IL-2 or modified IL-2; Discontinued trails |
|||
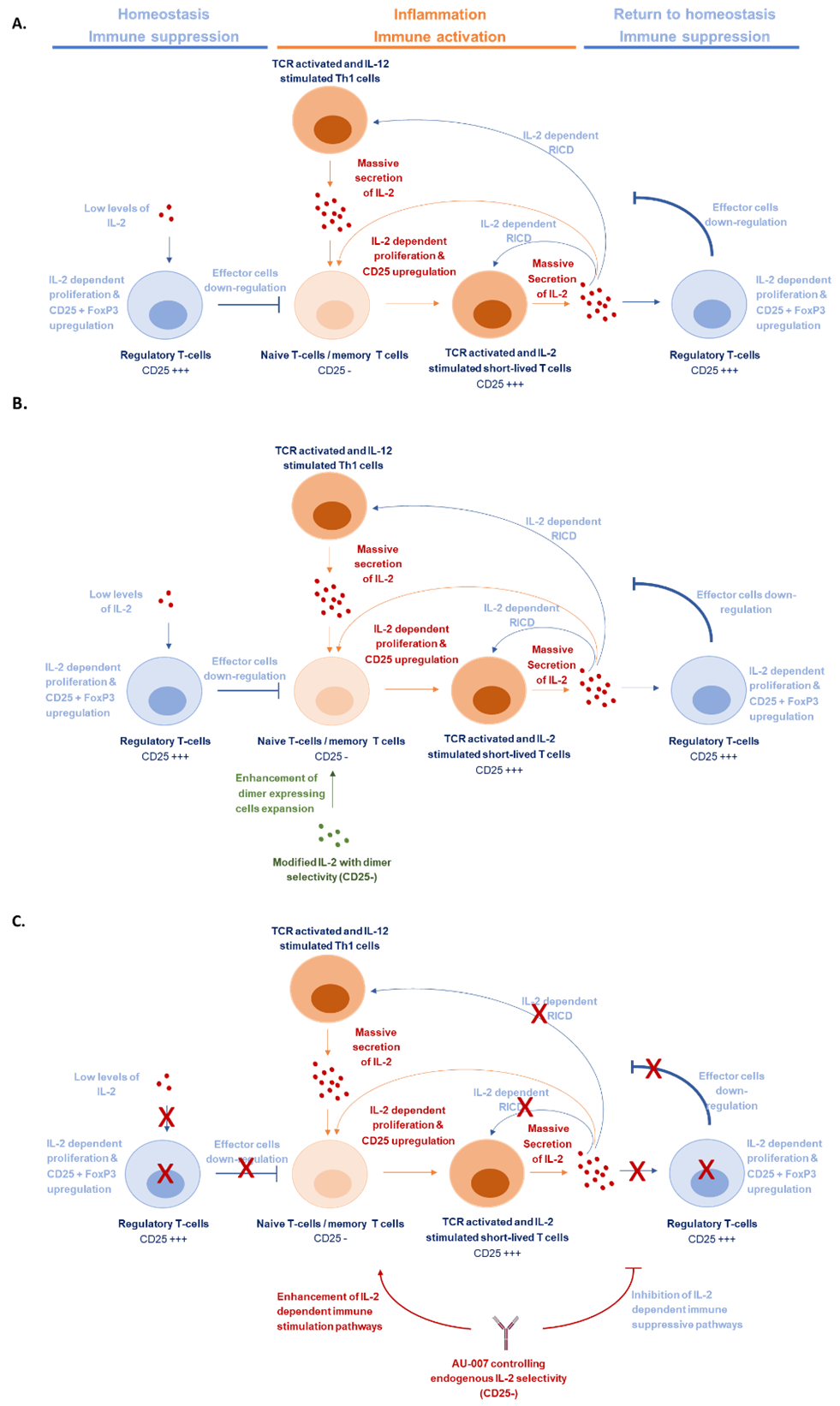
Figure 3. IL-2 negative feedback loop caused by endogenous IL-2 limits the activity of modified IL-2-based therapies. A. Schematic representation of IL-2 role as an immunomodulator in homeostasis and inflammation. B. Exogenous administration of modified IL-2 with bias selectivity to dimer-expressing cells promotes the expansion of CD25 negative (CD25-) effector cells yet is undermined by the endogenous IL-2 that pushes the system back to homeostasis. C. AU-007 captures and redirects endogenous IL-2, allowing it to expand CD25 negative (CD25-) effector cells while breaking the auto-inhibitory loop and expanding the inflammation & immune stimulation stage.
Conclusion
IL-2-based therapy can lead to significant responses in some cancer patients, but it is associated with significant toxicity. Modified IL-2 immune cytokines showed a cooperative yet limited anti-tumor effect, and recently suffered from setbacks in clinical trials. We propose that exogenous administration of IL-2 cytokines is inherently undermined by endogenously secreted IL-2. Immune activation by IL-2 is under tight negative feedback regulation through the IL-2-induced IL-2 release mechanism. While exogenous IL-2-based therapies cannot prevent Tregs expansion that is driven by the endogenous IL-2, controlling endogenous IL-2 by AU-007 enhances immune stimulation and cancer clearance.
Conflicts of Interest
The authors declare that the research presented in this paper was conducted by Biolojic Design Ltd. and was fully funded by Aulos Bioscience Inc. All authors are employed by, and have a personal financial interest in, either Biolojic Design Ltd. or Aulos Bioscience Inc. Biolojic-Design authors have stock options in Biolojic-Design. Aulos Bioscience authors have stock options in Aulos Bioscience.
Funding Statement
This work was sponsored by Aulos Bioscience Inc.
Author Contributions
I.A., N.L., M.G., M.B.M., T.W., and R.B., conceived the studies and analyzed and interpreted the data. I.A., N.L., M.G., M.B.M., R.B., and Y.O. drafted the manuscript. I.A., N.L., M.G., M.B.M., T.W., R.B., Y.S., S.F., J.V., A.K., R.H., and Y.O., reviewed the manuscript. S.F., G.N., and M.Z., computationally designed AU-007. S.F. computationally designed naIL-2. R.B., L.D., M.I., Y.F., production of naIL-2. N.L., M.G., and M.B.M., performed the in vitro experiments. L.D., and M.I., performed the complex detection ELISA study. O.B., and I.M., performed the surface plasmon resonance experiments.
Acknowledgments
We thank the following contributors for providing support: Biolojic Design team & Aulos Bioscience team for the great technical and scientific support.
References
2. Klatzmann D, Abbas AK. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nature Reviews Immunology. 2015 May;15(5):283-94.
3. Ross SH, Cantrell DA. Signaling and function of interleukin-2 in T lymphocytes. Annual Review of Immunology. 2018 Apr 26; 36:411-33.
4. Shatrova AN, Mityushova EV, Vassilieva IO, Aksenov ND, Zenin VV, Nikolsky NN, et al. Time-dependent regulation of IL-2R α-chain (CD25) expression by TCR signal strength and IL-2-induced STAT5 signaling in activated human blood T lymphocytes. PLoS One. 2016 Dec 9;11(12):e0167215.
5. Ballesteros-Tato A. Beyond regulatory T cells: the potential role for IL-2 to deplete T-follicular helper cells and treat autoimmune diseases. Immunotherapy. 2014 Nov;6(11):1207-20.
6. Pohida K, Lake CM, Yee D, Snow AL. Restimulation-Induced Cell Death (RICD): Methods for Modeling, Investigating, and Quantifying RICD Sensitivity in Primary Human T Cells via Flow Cytometric Analysis. Bio-Protocol. 2022 Feb 20;12(4):e4326.
7. Höfer T, Krichevsky O, Altan-Bonnet G. Competition for IL-2 between regulatory and effector T cells to chisel immune responses. Frontiers in Immunology. 2012 Sep 5;3:268.
8. Au-Yeung BB, Smith GA, Mueller JL, Heyn CS, Jaszczak RG, Weiss A, et al. IL-2 modulates the TCR signaling threshold for CD8 but not CD4 T cell proliferation on a single-cell level. The Journal of Immunology. 2017 Mar 15;198(6):2445-56.
9. Briukhovetska D, Dörr J, Endres S, Libby P, Dinarello CA, Kobold S. Interleukins in cancer: from biology to therapy. Nature Reviews Cancer. 2021 Aug;21(8):481-99.
10. Ross SH, Cantrell DA. Signaling and function of interleukin-2 in T lymphocytes. Annual Review of Immunology. 2018 Apr 26;36:411-33.
11. Bendickova K, Fric J. Roles of IL-2 in bridging adaptive and innate immunity, and as a tool for cellular immunotherapy. Journal of Leucocyte Biology. 2020 Jul;108(1):427-37.
12. Pol JG, Caudana P, Paillet J, Piaggio E, Kroemer G. Effects of interleukin-2 in immunostimulation and immunosuppression. Journal of Experimental Medicine. 2020 Jan 6;217(1):e20191247.
13. Nelson BH. IL-2, regulatory T cells, and tolerance. The Journal of Immunology. 2004 Apr 1;172(7):3983-8.
14. Malek TR, Bayer AL. Tolerance, not immunity, crucially depends on IL-2. Nature Reviews Immunology. 2004 Sep 1;4(9):665-74.
15. Oyler-Yaniv A, Oyler-Yaniv J, Whitlock BM, Liu Z, Germain RN, Huse M, et al. A tunable diffusion-consumption mechanism of cytokine propagation enables plasticity in cell-to-cell communication in the immune system. Immunity. 2017 Apr 18;46(4):609-20.
16. Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. The Journal of Immunology. 2014 Jun 15;192(12):5451-8.
17. Lenardo M, Chan FK, Hornung F, McFarland H, Siegel R, Wang J, et al. Mature T lymphocyte apoptosis—immune regulation in a dynamic and unpredictable antigenic environment. Annual Review of Immunology. 1999 Apr;17(1):221-53.
18. Richter GH, Mollweide A, Hanewinkel K, Zobywalski C, Burdach S. CD25 blockade protects T cells from activation-induced cell death (AICD) via maintenance of TOSO expression. Scandinavian Journal of Immunology. 2009 Sep;70(3):206-15.
19. Moon BI, Kim TH, Seoh JY. Functional modulation of regulatory T cells by IL-2. PLoS One. 2015 Nov 3;10(11):e0141864.
20. Choudhry H, Helmi N, Abdulaal WH, Zeyadi M, Zamzami MA, Wu W, et al. Prospects of IL-2 in cancer immunotherapy. BioMed Research International. 2018 Oct;2018:9056173.
21. Ahmadzadeh M, Rosenberg SA. IL-2 administration increases CD4+ CD25hi Foxp3+ regulatory T cells in cancer patients. Blood. 2006 Mar 15;107(6):2409-14.
22. Overwijk WW, Tagliaferri MA, Zalevsky J. Engineering IL-2 to give new life to T cell immunotherapy. Annual Review of Medicine. 2021 Jan 27;72:281-311.
23. Majidpoor J, Mortezaee K. Interleukin-2 therapy of cancer-clinical perspectives. International Immunopharmacology. 2021 Sep 1;98:107836.
24. Kalia V, Sarkar S. Regulation of effector and memory CD8 T cell differentiation by IL-2—a balancing act. Frontiers in Immunology. 2018 Dec 20;9:2987.
25. Shevyrev D, Tereshchenko V. Treg heterogeneity, function, and homeostasis. Frontiers in Immunology. 2020 Jan 14;10:3100.
26. Liu Z, Gerner MY, Van Panhuys N, Levine AG, Rudensky AY, Germain RN. Immune homeostasis enforced by co-localized effector and regulatory T cells. Nature. 2015 Dec 10;528(7581):225-30.
27. Jiang T, Zhou C, Ren S. Role of IL-2 in cancer immunotherapy. Oncoimmunology. 2016 Jun 2;5(6):e1163462.
28. Singh AV, Varma M, Laux P, Choudhary S, Datusalia AK, Gupta N, et al. Artificial intelligence and machine learning disciplines with the potential to improve the nanotoxicology and nanomedicine fields: a comprehensive review. Archives of Toxicology. 2023 Mar 7;97:963-79.
29. Zhou P. Emerging mechanisms and applications of low-dose IL-2 therapy in autoimmunity. Cytokine & Growth Factor Reviews. 2022 Oct;67:80-88.
30. Shatrova AN, Mityushova EV, Vassilieva IO, Aksenov ND, Zenin VV, Nikolsky NN, et al. Time-dependent regulation of IL-2R α-chain (CD25) expression by TCR signal strength and IL-2-induced STAT5 signaling in activated human blood T lymphocytes. PLoS One. 2016 Dec 9;11(12):e0167215.
31. Létourneau S, Krieg C, Pantaleo G, Boyman O. IL-2–and CD25-dependent immunoregulatory mechanisms in the homeostasis of T-cell subsets. Journal of Allergy and Clinical Immunology. 2009 Apr 1;123(4):758-62.
32. Dai Z, Arakelov A, Wagener M, Konieczny BT, Lakkis FG. The role of the common cytokine receptor γ-chain in regulating IL-2-dependent, activation-induced CD8+ T cell death. The Journal of Immunology. 1999 Sep 15;163(6):3131-7.
33. Bachmann MF, Oxenius A. Interleukin 2: from immunostimulation to immunoregulation and back again. EMBO Reports. 2007 Dec;8(12):1142-8.
34. Schmitz I, Krueger A, Baumann S, Schulze-Bergkamen H, Krammer PH, Kirchhoff S. An IL-2-dependent switch between CD95 signaling pathways sensitizes primary human T cells toward CD95-mediated activation-induced cell death. The Journal of Immunology. 2003 Sep 15;171(6):2930-6.
35. Dolgin E. IL-2 upgrades show promise at ASCO. Nature Biotechnology. 2022 Jul;40(7):986-988.
36. Raeber ME, Sahin D, Karakus U, Boyman O. A systematic review of interleukin-2-based immunotherapies in clinical trials for cancer and autoimmune diseases. EBioMedicine. 2023 Apr 1;90:104539.
37. Tanaka A, Sakaguchi S. Targeting Treg cells in cancer immunotherapy. European Journal of Immunology. 2019 Aug;49(8):1140-6.
38. Hu W, Wang G, Huang D, Sui M, Xu Y. Cancer immunotherapy based on natural killer cells: current progress and new opportunities. Frontiers in Immunology. 2019 May 31;10:1205.
39. Oh DY, Fong L. Cytotoxic CD4+ T cells in cancer: Expanding the immune effector toolbox. Immunity. 2021 Dec 14;54(12):2701-11.
40. Jiang T, Zhou C, Ren S. Role of IL-2 in cancer immunotherapy. Oncoimmunology. 2016 Jun 2;5(6):e1163462.