Commentary
Since the discovery of escaping mechanism of tumor from negative immune regulation, the paradigm of drug discovery for anti-cancer agents has been dramatically shifted to cancer immunotherapy (e.g., dendritic cell therapy, CAR-T cell therapy, or antibody therapy) by stimulating patient’s immune system to treat cancer [1-3]. Particularly, immune checkpoint inhibitors targeting PD-1, PD-L1, and CTLA-4 prevent tumor cells from evading immune surveillance and result in anti-tumor responses [4]. These immunomodulators were considered as a ‘game-changer’ with remarkable clinical achievement [5]. However, the response rate for immune checkpoint inhibitors was still variable and even quite low in a certain type of cancer despite their promising clinical outcome [6]. To explain the low response rate and compromised immune system, diverse biological processes are investigated for spontaneous anti-tumor immune response, but we aim to discuss T cell-inflamed tumor microenvironment in this commentary.
Immunophenotype of solid tumors is classified with two statuses by tumor microenvironment regulatory pathway – T cell-inflamed phenotype (‘hot’ tumor) versus non-T cellinflamed phenotype (‘cold’ tumor) [7]. In T cell-inflamed ‘hot’ tumors, type I interferon (IFN) signaling mediates expression of various chemokines and CD8+ DC lineage facilitates infiltration of CD8+ T cell [8]. In the clinical stage, patients with tumor-infiltrating T cell appeared as an activation of spontaneous immune response and a benefit with immunotherapy, whereas cancer recurrence was detected in patients with a lack of T cell infiltration [9,10]. Therefore, infiltration of T cells is a prognostic hallmark for positive clinical outcomes and indispensable for a desired therapeutic effect of immune checkpoint inhibitors. Type I IFN-mediated immune activation is one of the essential parts in the regulation of T cell-infiltration to guide tumor from ‘cold’ to ‘hot’.
IFNs are produced by activation of the innate immune system and mainly controlled anti-viral effect. Additionally, extensive studies suggested the therapeutic effect of IFN as an anti-cancer therapy. Even though both type I and II IFNs are important to promote anti-tumor immunity, only type I IFNs (IFNα and IFNβ) have been shown a favorable clinical outcome and approved for the treatment of cancer [11]. In cancer treatment, type I IFNs are usually used in hematological malignancy such as leukemia, sarcoma, melanoma, and lymphomas. IFNs are mainly injected by PEGylated form to improve clinical effect and stability, however still limited access to particular patients such as hyperbilirubinemia [12]. For that reason, the continuous efforts to develop small-molecule based-IFN inducers have been encouraged in the field of drug discovery.
To facilitate type I IFN signaling for anti-tumor responses, STING (Stimulator of interferon genes) has emerged as an innate immune regulator for the last decade [13]. At first, STING is known to play an important role in type I IFN-mediated innate immunity and anti-viral responses by sensing of DNA virus or self DNA [14,15]. STING is an ER-resident protein and activation of STING by ligand binding leads to translocate from ER to Golgi/ER-Golgi intermediate compartment for recruiting downstream kinase TBK1 [16]. Then, activation of TBK1 promotes signaling cascade for STING-TBK1-IRF3, followed by phosphorylation of STING and IRF3, then dimerization of phosphor-IRF3 to trigger IFNB gene transcription, and eventually resulted in activation of type I IFN signaling [16]. Based on the critical role of type I IFN signaling in host immune surveillance of cancer, IFNβ production by activation of the host STING pathway induced spontaneous T cell response and tumor regression, which means STING-mediated type I IFN phenotype as a key mechanism of action for targeting non-T cell-inflamed tumor [17,18]. Moreover, pharmacological STING activation demonstrated a positive therapeutic effect to overcome the current hurdle for drug-resistance cancer [19,20]. Therefore, the activation of STING pathway has been spotlighted for the next generation of cancer immunotherapy.
For the discovery of natural ligands for STING, Burdette et al. [21] firstly reported that cyclic diguanylate (c-di- GMP) from bacteria directly binds STING as a sensor of cyclic dinucleotide (CDN). Since the discovery of c-di- GMP as a second messenger for innate immune response mediated by STING, subsequent studies demonstrated that STING directly recognized not only the bacterial cyclic dinucleotide (CDN) such as c-di-AMP (cyclic diadenylate) or 3,3-cGAMP (Cyclic GMP-AMP) but also the mammalian CDN, 2,3-cGAMP produced by cGAS (Cyclic GMP-AMP synthase) [22-24]. These results led to the development of synthetic CDN derivatives as a new anti-cancer drug for immunotherapy by promoting spontaneous immune response of non-T cell-inflamed tumor. The first synthetic human STING agonist is ADU-S100 (Figure 1), which is mimetic of CDN with alternative phosphate bridge and ADU-S100 remarkably improved anti-tumor efficacy compared to 2,3-cGAMP and suppressed re-challenge of the same tumor [17,18]. Based on these features, ADU-S100 is now on phase 2 clinical trial targeting metastatic and recurrent head and neck cancer combined with immune checkpoint inhibitors [25]. Nonetheless, CDN derivatives are limited in clinical study due to narrow application of tumors because they were mostly administrated by intratumoral (IT) injection [20]. To address these issues, there exist continuous effort for the development of non- CDN based STING agonists.
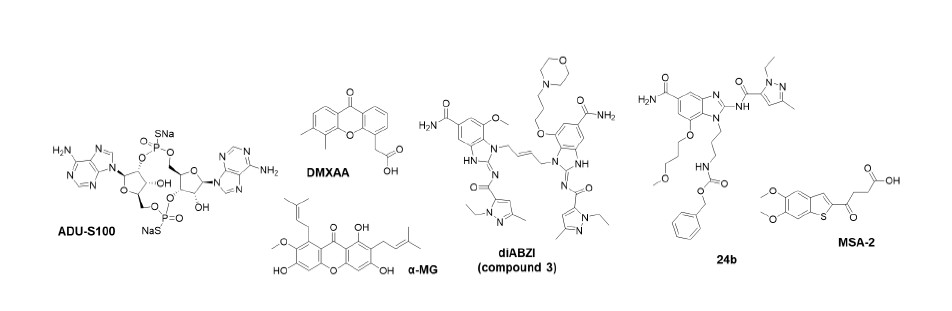
DMXAA (also known as ASA404 and Vadimezan, Figure 1) bearing xanthene structure has been known to produce an anti-tumor response in mouse model causing vascular disruption, however, ultimately failed in human clinical trials [26]. Further mechanistic investigation for DMXAAbinding target elucidated that DMXAA is a murine-specific STING agonist, thereby the treatment of DMXAA for type I IFN immunity is only sensitive for mouse STING, not human STING [27,28]. With a structural insight for DMXAA, Zhang et al. discovered a natural xanthaonoid, α-Mangostin (α-MG, Figure 1) binds and stabilized human STING C-terminal domain to activate TBK1-IRF3 cascade, followed by type I IFN signaling, but binding affinity and potency of α-MG are quite controversial compared to cGAMP [29].
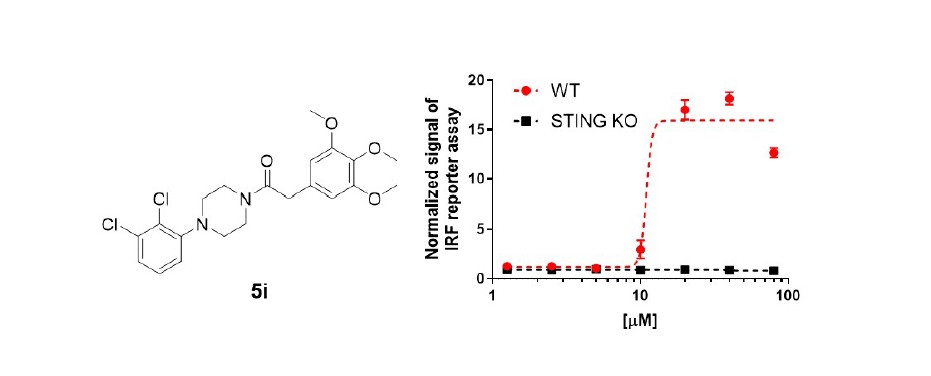
For anti-tumor immunity, the first non-CDN STING agonist embedded with amidobenzimidazole as a pharmacophore was reported by Ramanjulu et al. [30]. Based on the structure information of STING, the design strategy for synthetic STING agonists is two symmetrylinked amidobenzimidazole (diABZI) introducing dimerization of core skeleton (Figure 1). As a result, diABZI dramatically improved the binding affinity and efficacy for STING. diABZI (compound 3) is 400-fold more potent than cGAMP which demonstrated inhibition of tumor growth and improvement of tumor-bearing mice survival by intravenous (i.v.) injection [30]. Moreover, the study of the structure-activity relationship for ABZI monomer was reported for STING agonistic effect [31]. Compound 24b with N-carbobenzyloxy aminopropyl moiety (Figure 1) effectively activated STING and type I IFN signaling as a monomeric form, thereby significantly reduced tumor growth even with 0.15 mpk i.v. administration [31]. On the other hand, the approach targeting much ‘small’ molecule discovered a new orally available STING agonist, MSA-2, by phenotypic screening for chemical inducers of IFNβ secretion (Figure 1) [32]. In the case of MSA-2, the monomer is not available to bind STING, whereas the non-covalent pre-dimerization of MSA-2 induced STING binding and activation [32]. Besides, MSA-2 can accumulate in tumor tissue due to an acidic tumor microenvironment, which allowed the systematic administration of STING agonist [32]. Recently, we reported a study of a new IFN-inducing compound, 5i with arylpiperazine as a pharmacophore (Figure 2) that effectively activated IFNβ secretion and type I IFN immune response in THP-1 human monocyte cells [33]. By further verification, we demonstrated 5i-mediated immune response appealed in a STINGdependent manner (Figure 2). These results suggested the potential ability of a new core skeleton for small-molecule STING agonists.
Conclusion
The therapeutic effect of small-molecules stimulating type I IFN phenotype is now considered to open a new area for cancer immunotherapy. Especially, activation of STING pathway is extremely important for the regulation of type I IFN immune response and anti-cancer effect promoting spontaneous T cell response. The approach developing small-molecules for STING agonists may lead to a shift of drug discovery paradigm in cancer therapy to overcome a low response rate against immune checkpoint inhibitors and cure non-T cell-inflamed tumors.
Acknowledgments
Authors acknowledge financial supports of Bio & Medical Technology Development Program (NRF- 2019M3E5D4066905) and Priority Research Centers Program (2019R1A6A1A11051471) of the National Research Foundation (NRF) funded by Korean government (MSIT).
References
2. Palucka K, Banchereau J. Dendritic-Cell-Based Therapeutic Cancer Vaccines. Immunity. 2013;39:38–48.
3. Waldmann TA. Immunotherapy: Past, present and future. Nat Med. 2003;9:269–77.
4. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015;33:1974–82.
5. Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat Commun. 2020;11:10–2.
6. Ganesan S, Mehnert J. Biomarkers for Response to Immune Checkpoint Blockade. Annu Rev Cancer Biol. 2020;4:331–51.
7. Trujillo JA, Sweis RF, Bao R, Luke JJ. T cell–inflamed versus Non-T cell–inflamed tumors: a conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol Res. 2018;6:990–1000.
8. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–22.
9. Galon J, Pagès F, Marincola FM, Angell HK, Thurin M, Lugli A, et al. Cancer classification using the Immunoscore: A worldwide task force. J Transl Med. 2012;10.
10. Larson HM. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict. Jay Cooke, Priv Bank. 2014;313:437–98.
11. Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836–48.
12. Zwirtes R, Narasimhan P, Wind-Rotolo MM, Xu D, Hruska MW, Kishnani N, et al. Mechanisms of hyperbilirubinemia during peginterferon lambda-1a therapy for chronic hepatitis c infection: A retrospective investigation. J Interf Cytokine Res. 2016;36:644–51.
13. Sheridan C. Drug developers switch gears to inhibit STING. Nat Biotechnol. 2019;37:199–201.
14. Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci U S A. 2012;109:19386–91.
15. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–42.
16. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. (80- ) 2015;347.
17. Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015;11:1018–30.
18. Sivick KE, Desbien AL, Glickman LH, Reiner GL, Corrales L, Surh NH, et al. Magnitude of Therapeutic STING Activation Determines CD8+ T Cell-Mediated Anti-tumor Immunity. Cell Rep. 2018;25:3074-3085.e5.
19. Chipurupalli S, Ganesan R, Dhanabal SP, Kumar MS, Robinson N. Pharmacological STING Activation Is a Potential Alternative to Overcome Drug-Resistance in Melanoma. Front Oncol. 2020;10:1–9.
20. Cheng N, Watkins-Schulz R, Junkins RD, David CN, Johnson BM, Montgomery SA, et al. A nanoparticleincorporated STING activator enhances antitumor immunity in PD-L1-insensitive models of triple-negative breast cancer. JCI Insight .2018;3:1–20.
21. Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–8.
22. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, et al. CGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380–4.
23. Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, et al. Structure-function analysis of STING activation by c[G(2',5') pA(3',5')p] and targeting by antiviral DMXAA. Cell. 2013;154:748–62.
24. Kato K, Omura H, Ishitani R, Nureki O. Cyclic GMPAMP as an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. (80- ) 2013;339:826–30.
25. Naour J Le, Zitvogel L, Galluzzi L, Vacchelli E, Le J, Zitvogel L, et al. Trial watch : STING agonists in cancer therapy. Oncoimmunology. 2020;9.
26. Lara PN, Douillard JY, Nakagawa K, Von Pawel J, McKeage MJ, Albert I, et al. Randomized phase III placebo-controlled trial of carboplatin and paclitaxel with or without the vascular disrupting agent vadimezan (ASA404) in advanced non-small-cell lung cancer. J Clin Oncol. 2011;29:2965–71./a>
27. Conlon J, Burdette DL, Sharma S, Bhat N, Thompson M, Jiang Z, et al. Mouse, but not Human STING, Binds and Signals in Response to the Vascular Disrupting Agent 5,6-Dimethylxanthenone-4-Acetic Acid. J Immunol. 2013;190:5216–25.
28. Kim S, Li L, Maliga Z, Yin Q, Wu H, Mitchison TJ. Anticancer flavonoids are mouse-selective sting agonists. ACS Chem Biol. 2013;8:1396–401.
29. Zhang Y, Sun Z, Pei J, Luo Q, Zeng X, Li Q, et al. Identification of a-Mangostin as an Agonist of Human STING. ChemMedChem. 2018;13:2057–64.
30. Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang SY, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. 2018;564:439–43.
31. Xi Q, Wang M, Jia W, Yang M, Hu J, Jin J, et al. Design, Synthesis, and Biological Evaluation of Amidobenzimidazole Derivatives as Stimulator of Interferon Genes (STING) Receptor Agonists. J Med Chem. 2020;63:260–82.
32. Pan BS, Perera SA, Piesvaux JA, Presland JP, Schroeder GK, Cumming JN, et al. An orally available nonnucleotide STING agonist with antitumor activity. Science. 2020;369(6506):eaba6098.
33. Chu Y, Reddy BR, Gajulapalli VP, Babu KS, Kim E, Lee S. Design, synthesis, and biological evaluation of N-arylpiperazine derivatives as interferon inducers. Bioorganic & Medicinal Chemistry Letters. 2020 Dec 15;30(24):127613.