Abstract
Staphylococcus aureus is a non-motile Gram-positive bacterium that exhibits antibiotic resistant forms (i.e. MRSA, VRSA), which continue to represent a major source of nosocomial infection. Alternative therapeutics are urgently needed to control the overactive host response in S. aureus-associated sepsis. However, the therapeutic molecular targets for sepsis remain poorly understood. We aim to identify these molecular targets to better understand the complex host response and to spur the development of novel therapeutics. MKP-1 plays a critical role in negatively regulating S. aureus-induced inflammatory cytokine production in macrophages. We found that S. aureus can induce MKP-1 expression in vitro. Knocking out MKP-1 results in significantly higher levels of TNFα and IL-1β gene expression during S. aureus infection of bone marrow-derived macrophages (BMDMs). Pharmacological induction of MKP-1 by rolipram produces significantly decreased levels of TNFα. The mechanism of MKP-1 expression by S. aureus requires Toll-like receptor 2 (TLR-2). Together, these findings indicate that MKP-1 is a negative regulator during S. aureus infection.
Keywords
MKP-1, DUSP-1, S. aureus, Sepsis, Inflammatory cytokines, TNFα
Introduction
Sepsis continues to be a major obstacle for the US healthcare system, with approximately 660,000 cases reported in 2000 [1]. An analysis of health data collected by the Centers for Disease Control and Prevention showed that the US experienced 240,000 deaths from sepsis in 2022 [2]. The overall age adjusted mortality rate for sepsis was found to be 89 per 100,000 people [2]. Treatment consists of broad-spectrum antibiotics; however, antibiotic-resistant infections represent a significant cause of sepsis [3]. Therefore, novel therapeutics are urgently needed to control the overactive host response in sepsis.
Approximately half of all bacterial sepsis cases are due to infection by a Gram-positive bacterium [4]. Of these cases, 20.5% result from Staphylococcus aureus infection; an additional 10.2% are due to Methicillin resistant S. aureus (MRSA) [4]. Another antibiotic resistant strain is Vancomycin resistant S. aureus, which represents a growing cause of nosocomial infection [5,6]. S. aureus is a non-motile Gram-positive bacterium that is responsible for causing pneumonia, skin and soft tissue infections, endocarditis, osteomyelitis, and sepsis. During sepsis, S. aureus induces Toll-like receptor 2 (TLR-2) to provoke an exaggerated inflammatory response [7]. In the innate immune system, TLR-2 functions as a pattern recognition receptor against common bacterial antigens [7]. Activation of TLR-2 leads to downstream production of inflammatory cytokines, such as TNFα and IL1-β [8].
Inflammation is necessary for clearing bacterial infections, but it can be detrimental if left unregulated. Therefore, termination of inflammation is a tightly controlled part of the host immune response [9]. Mitogen-activated protein kinase phosphatase-1 (MKP-1) is a critical negative regulator of inflammation [10,11]. Both proinflammatory cytokines (e.g., TNFα) and MKP-1 are expressed in response to TLR activation [10]. Proinflammatory cytokines cause inflammation by stimulating immune function, while MKP-1 dampens the immune response by inhibiting proinflammatory mediators (i.e., p38, ERK, and JNK) [12,13]. In vitro studies have shown that increased TNFα leads to greater MKP-1 expression, which allows for a carefully regulated immune response [13]. However, this negative feedback system is less effective against S. aureus infection, which can induce an exaggerated inflammatory response that is unaffected by host anti-inflammatory mechanisms. In our study, we aim to determine if S. aureus can elicit MKP-1 production in vivo.
Rolipram is a phosphodiesterase 4 (PDE-4) inhibitor which has been shown to upregulate MKP-1 expression in mouse macrophage cell-lines treated with lipopolysaccharide (LPS) [14,15]. PDE-4 inhibitors selectively degrade secondary messengers of the inflammatory response, such as cAMP [16]. Rolipram induced upregulation of MKP-1 was associated with a dose-dependent decrease in TNFα [14,15].
Based on previously published work, we hypothesize that MKP-1 plays a similar role in an in vivo S. aureus infection model. We performed experiments to demonstrate that S. aureus can induce MKP-1 expression in RAW 264.7 macrophages and primary bone marrow-derived macrophages (BMDMs). To investigate the function of MKP-1 in macrophages after S. aureus infection, we verified that the absence of MKP-1 impacts the expression of proinflammatory genes. Additionally, the pharmacological induction of MKP-1 negatively regulates TNFα transcription in macrophages after S. aureus infection. Finally, we demonstrated that TLR-2 is responsible for initiating signaling mechanisms that ultimately lead to MKP-1 induction.
Materials and Methods
Bacterial strains and culture
A clinical isolate of S. aureus strain USA300 was generously provided by Dr. Randall Worth and was characterized as previously described [17]. Escherichia coli K12 (substrain W3110; ATCC 27325) was generously provided by Dr. R. Mark Wooten. Bacteria were cultured in Bacto™ Tryptic Soy Broth (Becton, Dickinson and Company; Sparks, MD) in a 250mL sterile, plain bottom, vented closure PETG Nalgene flask (Thermo Scientific; Rochester, NY) overnight at 37°C and 200 RPM.
Bacterial quantitation
Overnight culture was pelleted, suspended in 1x DPBS (Life Technologies; Grand Island, NY), and absorbance was read at OD:600 using a SmartSpec™ 3000 (Bio-Rad; Hercules, CA). Absorbance of 0.8 at OD:600, whereby S. aureus = (2x10^8 CFU/mL) and E. coli = (6x10^8 CFU/mL).
Mice
Mice were maintained in the animal facilities of the University of Toledo Health Science Campus. All experiments were performed in accordance with and approved by the University of Toledo IACUC. Wildtype and MKP-1 knockout C57BL/6 mice were purchased from the NCI mouse repository (Frederick, MD).
Cell culture
RAW 264.7 macrophages were cultured in DMEM (1x)+GlutaMAX™-I media (GIBCO, Life Technologies; Grand Island, NY) containing 4.5 g/L D-Glucose, without sodium pyruvate and supplemented with 10% US Qualified FBS Lot # 1365352 (GIBCO, Life Technologies; Grand Island, NY) and 1% Penicillin-Streptomycin (5,000 U/mL) (GIBCO, Life Technologies; Grand Island, NY). Primary bone marrow-derived macrophages (BMDMs) were differentiated and isolated as previously described [18]. RAW and primary macrophages were seeded in culture media overnight prior to experimentation.
Macrophage infection assay
Macrophages were seeded into 6-well or 24-well tissue culture treated plates at a final concentration of 2.00x106 per well or 2.00x105 per well, and they were allowed to adhere overnight at 37ºC in a 5% ambient CO2 incubator. Media was removed from the plated macrophages and bacteria was added at an MOI of 100. The plates were then gently centrifuged at 300g for 5 minutes, then returned to a CO2 incubator for 1 hour to allow internalization of bacteria. After 1 h the cells were washed twice with room temperature 1x DPBS (Life Technologies; Grand Island, NY) to remove any extracellular bacteria. After the second wash, fresh pre-warmed media was added to the macrophages for the duration of the experiment, and the cells were returned to the incubator. At each experimental time point, cells were washed once with 4°C 1x DPBS to remove any residual media. Cells were suspended in 100 µL 4°C 1x DPBS and lysed with RIPA Lysis and Extraction buffer (Pierce, Thermo Scientific; Rockford, IL) supplemented with 1x halt protease and phosphatase inhibitor cocktail (Pierce, Thermo Scientific; Rockford, IL) for western blot samples, or RLT buffer for qPCR samples. The samples were vortexed vigorously and stored at -80°C for later analysis.
Ligand stimulation of macrophages
Peptidoglycan (PGN) (InvivoGen; San Diego, California) and lipoteichoic acid (LTA) (InvivoGen; San Diego, California) derived from S. aureus were suspended according to the manufactures recommended instructions and stored at a final concentration of 200 µg/mL or 5 mg/mL aliquots respectively at -20°C until use. Peptidoglycan or lipoteichoic acid were added to macrophages.
Rolipram treatment of macrophages
Rolipram (MP Biomedicals, LLC; Illkirch, France) was suspended according to the manufactures recommended instructions and stored at -20°C until use. For each experiment an aliquot was thawed and suspended in pre-warmed experimental media to a final concentration ranging from 0.1-100 µM.
Propidium iodide assay
Macrophages were infected as previously described [14]. Cells were stained with propidium iodide nucleic acid stain (Molecular Probes, Life Technologies; Grand Island, NY) following the manufactures protocol and florescence read on a Tali® instrument. The ratio of non-fluorescent to fluorescent cells was used to determine percent viability.
Total internal reflection fluorescence microscopy
Raw 264.7 Cells were seeded at 2x105 cells per well in 2 mL volumes on 24 mm round cover glass (Fisher Scientific; Pittsburgh, PA), within clear plastic 6-well tissue culture plates (Corning; Fisher Scientific; Pittsburgh, PA) and incubated overnight at 37°C in 5% CO2. The following day, macrophages adhering to coverslips were infected with S. aureus and incubated at 37°C in 5% CO2 for 1 h. After incubation, the coverslip was washed three times twice with room temperature 1x DPBS (Life Technologies; Grand Island, NY) aspirating between washes. The cover slip was placed on a mounting plate and 1 mL of fresh pre-warmed experimental media was added. The mounting plate and coverslip were then placed within an environmental chamber (Live Cell, Pathology Devices Inc, Westminster, MD) that maintained cells at 37°C, humidified, and contained 5% CO2 atmosphere. Bright-field images were acquired at 5 min intervals using a 40x, 1.35 numerical aperture (NA), Olympus objective on a Total Internal Reflection Fluorescence (TIRF) microscope (Olympus; Shinjuku, Tokyo). Images were processed using MetaMorph (for Olympus Premier image analysis software version 7.5.6.0) and exported as an AVI video file.
Western blot
The cell lysates were thawed on ice, cleared of cellular debris, and total protein quantified using the BCA protein assay (Pierce, Thermo Scientific; Rockford, IL). Sample were run Bolt® Bis-Tris plus 4-12% precast polyacrylamide gradient gels in 1x Bolt® MES SDS running buffer (Life Technologies; Grand Island, NY) at 165V for 36 min. Protein was transferred using the iBlot® gel transfer system (Life Technologies; Grand Island, NY ) and blocked for 1 h in Odyssey® blocking buffer (LI-COR; Lincoln, Nebraska). The membrane was incubated with primary antibody overnight, washed using 1x 0.2% DPBST, incubated for 1 h in IRDye secondary antibodies (LI-COR; Lincoln, Nebraska), washed three times, and then scanned using the Odyssey® infrared imaging system (LI-COR; Lincoln, Nebraska). Images were analyzed using Image studio V2.1 (LI-COR; Lincoln, Nebraska).
Quantitative polymerase chain reaction
Total mRNA was isolated with the RNeasy mini kit (Qiagen; Venlo, Limburg, Germany). Total mRNA quantity and purity was analyzed using a NanoDrop 2000 spectrophotometer (Thermo scientific; Rochester, NY). Reverse transcription was performed using the QuantiTect reverse transcription kit (Qiagen; Venlo, Limburg, Germany). cDNA was diluted 1:10 and added to FastStart essential DNA green master mix (Roche Diagnostics Corporation; Indianapolis, IN). qPCR was performed on the LightCycler96 (Roche Diagnostics Corporation; Indianapolis, IN). All qPCR data was analyzed using LightCycler96 software version 1.1.0.1320 (Roche Diagnostics Corporation; Indianapolis, IN).
Graphing and statistical analyses
Data was analyzed on a personal computer using GraphPad Prism version 5.03 for Windows (GraphPad Software, San Diego, CA, USA, www.graphpad.com). Statistical significance was determined using either GraphPad InStat version 3.06 for Windows (GraphPad Software, San Diego, CA), Microsoft Excel 2010 (Microsoft Corporation), or SPSS Statistics Version 21 (IBM; Armonk, NY). Datasets were evaluated using a one-way ANOVA with Tukey’s post-hoc test. P-values ≤ 0.05 were considered statistically significant, with experiments completed in triplicate.
Results
S. aureus can induce MKP-1 expression in macrophages.
We first verified the published literature showing that macrophage cell-lines upregulate MKP-1 in response to bacterial infection. Mouse RAW 264.7 macrophages were infected with S. aureus or E. coli for 0.5–4 h. Coinciding with previous findings, E. coli infection of RAW 264.7 macrophages induced MKP-1 protein expression after 0.5h from infection and expression was maintained through the 4 h time point (Figure 1A). S. aureus induced maximal MKP-1 expression at 4 h post infection (Figure 1A). MKP-1 induction was similar between cell-lines and primary BMDMs when infected with S. aureus or E. coli (Figure 1B). Taken together, the results confirm that MKP-1 is induced during bacterial infection of macrophage cell-lines and BMDMs. We were able to show similar kinetics of MKP-1 induction in both types of macrophages.
To alleviate the concern that an MOI of 100 may kill the macrophages, cell viability was monitored. Membrane integrity was not affected up to 6h post infection when examined by propidium iodide staining (Figure S1). Additionally, S. aureus infected RAW 264.7 macrophages were visualized by bright-field images collected using TIRF (Figure S1). Macrophages were found to be adherent after infection, although cell rounding was observed starting at 13–14 h post infection. Therefore, we focused on data acquired after a maximum of 4 h of infection to avoid potential nonspecific effects of cell toxicity on MKP-1 regulation and activity.
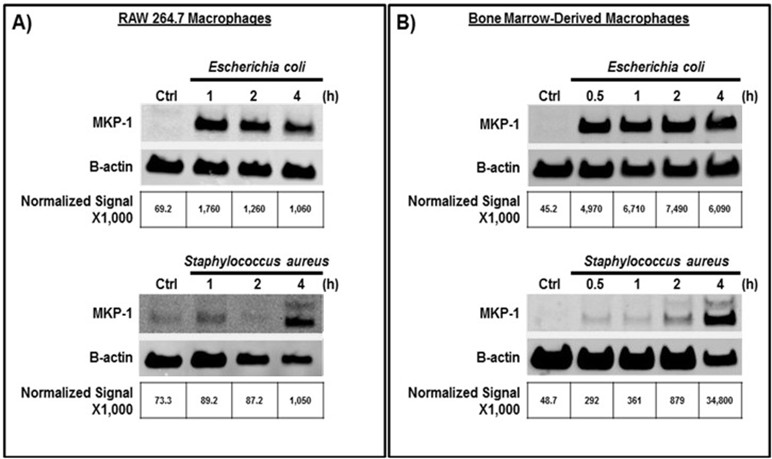
Figure 1. S. aureus can induce MKP-1 expression in RAW 264.7 macrophages and BMDMs from C57BL/6 mice. (A) RAW 264.7 cells or (B) BMDMs were infected with E. coli or S. aureus (MOI = 100). Macrophages were lysed at indicated times post-infection and western blot performed.
MKP-1 knockout BMDMs produce significantly higher levels of inflammatory gene expression during S. aureus infection
Next, we examined inflammatory cytokine production during S. aureus infection in macrophages lacking MKP-1. We infected BMDMs from wild type and MKP-1 knockout mice. We detected significantly higher levels of TNFα and IL-1β gene transcripts in MKP-1 knockout BMDMs at all-time points (Figures 2A and 2B). These results suggest that MKP-1 serves as a negative regulator of inflammatory cytokine production in vivo.
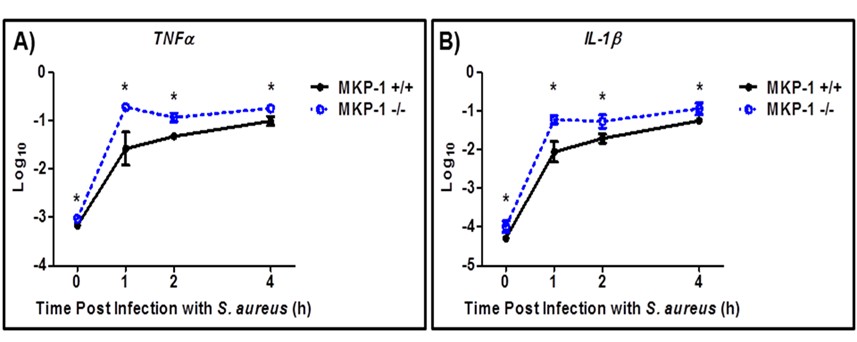
Figure 2. Knockout of MKP-1 results in significantly elevated (A) TNFα and (B) IL-1β mRNA expression during S. aureus infection (MOI 100). All significant values (*P ≤ 0.05) were compared to the wild-type at the same time point. Data shown as mean ± SEM (n=3 total); each experiment was performed in triplicate.
Pharmacological induction of MKP-1 significantly decreases TNFα expression in RAW 264.7 macrophages during S. aureus infection
We’ve previously shown that in S. aureus infected RAW 264.7 macrophages, pretreatment with 10 µM Rolipram was shown to induce MKP-1 [14]. Rolipram exhibited peak MKP-1 mRNA expression after a 1 h pretreatment [14]. In our current study, we pretreated RAW 264.7 macrophages with 0.1-100 μM rolipram for 1 h. This resulted in a dose-dependent increase in MKP-1 mRNA and a decrease in TNFα mRNA (Figure 3A). These findings suggest that S. aureus-induced TNFα production can be inhibited by rolipram pretreatment in a dose dependent manner, via the induction of MKP-1.
We observed a significant effect when we compared MKP-1 transcript levels in infected RAW 264.7 macrophages pretreated with 10 µM rolipram compared to uninfected pretreated macrophages (Figure 3B). This suggests the possibility of an additive effect on MKP-1 induction between rolipram (10 µM) and infection with S. aureus.
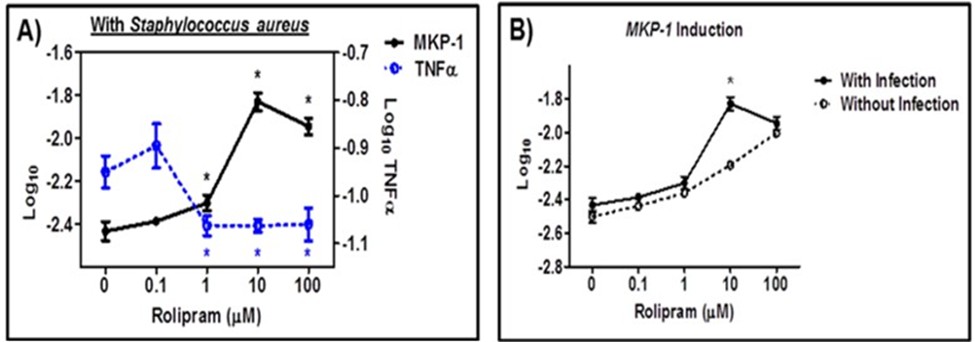
Figure 3. RAW 264.7 macrophages pretreated with Rolipram exhibited (A) increased MKP-1 mRNA expression, and decreased TNFα mRNA expression. (B) Rolipram-induced MKP-1 mRNA expression was increased during S. aureus infection (MOI 100). All significant values (*P ≤ 0.05) were compared to (A) 0 µM Rolipram or (B) similar doses with or without infection; each experiment was performed in triplicate.
TLR-2 ligands are sufficient for MKP-1 induction and TLR-2 is required for maximal MKP-1 induction by S. aureus
To evaluate if MKP-1 induction occurs through TLR signaling, we first determined whether TLR-2 ligands could elicit MKP-1 production. Lipoteichoic acid (LTA) and peptidoglycan (PGN) are two common TLR-2 ligands derived from S. aureus [19]. When treated with LTA and PGN for 1 h, BMDMs exhibited significantly increased MKP-1 mRNA expression (Figure 4A). This suggests that TLR-2 ligands derived from S. aureus are sufficient for MKP-1 induction.
To study these effects in the context of a live infection rather than purified subunits, we next compared S. aureus-induced MKP-1 production in BMDM derived from wild-type and TLR-2 deficient C57BL/6 mice. S. aureus was able to elicit MKP-1 induction in wild-type macrophages; however, in the absence of TLR-2 there was no induction of MKP-1 mRNA (Figure 4B). This data suggests that TLR-2 is required for S. aureus-induced MKP-1 expression in macrophages.
An alternative hypothesis is that MKP-1 induction by S. aureus is due to secreted bacterial factors rather than surface ligands. To evaluate this hypothesis, we infected BMDMs with heat-killed S. aureus. As with the live bacterial infection, heat-killed S. aureus induced MKP-1 production in wild-type, but not the TLR-2 knockout BMDMs (Figure 4C). In addition to MKP-1, we also examined TNFα induction in wild-type and TLR-2 knockout BMDMs. BMDMs infected with live or heat-killed bacteria produced increased TNFα mRNA in response to bacterial infection in both wild-type and TLR-2 knockout cells (Figures 4B and 4C).
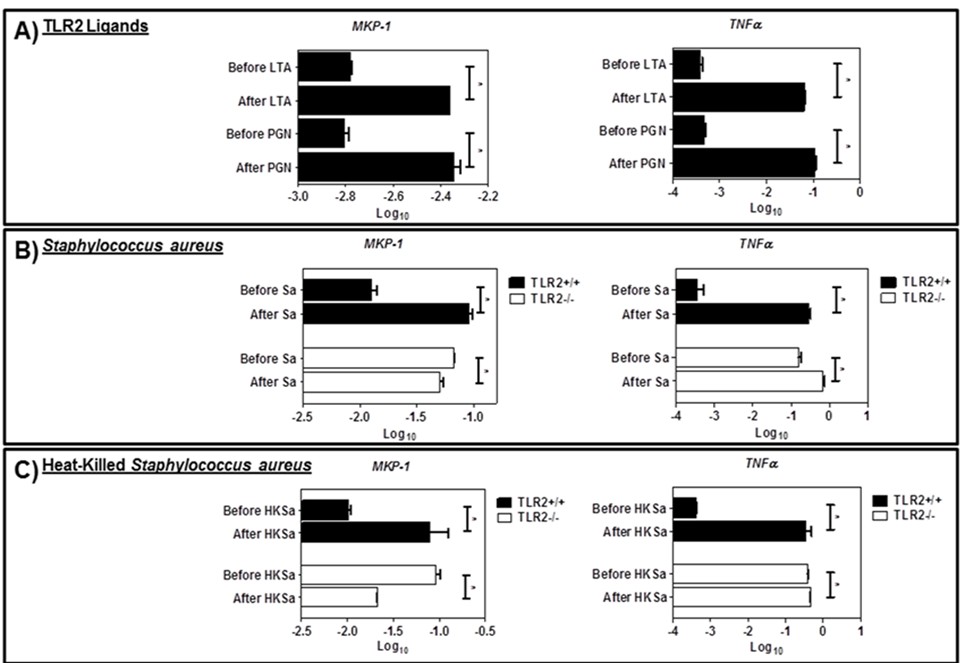
Figure 4. MKP-1 and TNFα mRNA expression was increased (A) in response to pretreatment with LTA and PGN. In wild-type BMDMs, (B,C) MKP-1 and TNFα were increased in response to live (Sa) and heat killed S. aureus (HKSa). In TLR-2 knockout BMDMs, (B,C) MKP-1 expression was decreased in response to Sa and HKSa infection. TNFα expression was minimally increased in response to Sa and HKSa infection. All significant values (*P ≤ 0.05) were compared to the wild-type at 4 h post infection. Data shown as mean ± SEM and are one representative experiment (n=3 total); each experiment was performed in triplicate.
Discussion
We determined that MKP-1 can be induced by S. aureus infection in RAW 267.4 cells and BMDMs. TLR-2 signaling is necessary for MKP-1 induction after S. aureus infection. Taken together, we have identified that MKP-1 may be an important target for future development of novel therapeutics for S. aureus associated sepsis. These medications are needed against quickly expanding drug-resistant S. aureus infections.
In our first aim, we determined that MKP-1 is induced by E. coli and S. aureus infection in RAW 264.7 macrophages. Previous work identified that MKP-1 can be elicited by macrophage cell-lines in response to Gram-negative bacteria [11]. However, it was unclear if Gram-positive bacteria also elicited MKP-1 in an in vivo setting. We demonstrated that both live and heat-killed S. aureus elicit MKP-1 mRNA expression in BMDMs. Interestingly, the maximal production of MKP-1 protein was observed 4 h after S. aureus infection. This is of note because MKP-1 has generally been described as an immediate early protein, reaching maximal expression in 15–60 min in vitro [10]. We showed maximal MKP-1 protein induction occurring 30 min after E. coli infection and at 4 h post infection with S. aureus. This may suggest that the kinetics by which macrophages regulate inflammatory cytokines (e.g., TNFα) are different depending on the infection. Also, our use of a 100 μM MOI infection may explain the delay in peak MKP-1 expression because higher titers will result in a larger initial inflammatory response. Future studies, such as a MKP-1 reporter assay, could determine differences in MKP-1 promoter activation in response to different bacterial infections. Taken together, we have identified that S. aureus can elicit MKP-1 production and that kinetics of production differ depending on the infection type.
In our second aim, we verified that MKP-1 functions as a negative regulator of S. aureus-induced inflammatory cytokines in BMDMs. By increasing MKP-1 expression using rolipram, we demonstrated that the increase coincided with a reciprocal decrease in TNFα mRNA levels. This suggests an inverse relationship between MKP-1 and inflammatory gene expression. We sought to further establish this relationship using a second line of evidence. Experiments using BMDMs derived from MKP-1 knockout mice illustrated that in the absence of MKP-1, higher levels of TNFα mRNA are produced. Additional studies using MKP-1 overexpressing cells can provide further evidence that MKP-1 downregulates inflammatory processes during S. aureus infection. By tempering the production of cytokine gene transcripts, MKP-1 likely has a role in preventing sepsis from S. aureus infections.
In our third aim, we determined that TLR-2 is necessary for MKP-1 induction after S. aureus infection in BMDMs. Previous studies have shown that TLRs are involved in MKP-1 expression, however, the requirement for specific receptors has not been well-established [20]. We demonstrated that TLR-2 ligands (i.e., PGN and LTA) are sufficient to induce MKP-1 mRNA expression. Using live or heat-killed S. aureus infection in wild-type and TLR-2 knockout BMDMs, we demonstrated that TLR-2 activation (i.e., via bacterial surface ligands) is necessary for maximal MKP-1 mRNA induction. Without TLR-2, BMDM’s were unable to upregulate MKP-1 in response to infection. In both TLR-2 wild-type and knockout BMDMs, TNFα was upregulated after infection. This suggests that while MKP-1 is reliant on TLR-2 activation, TNFα can be produced in response to activation of other pattern recognition receptors. Overall, the results of specific aim 3 suggest that TLR-2 ligands are sufficient for MKP-1 induction and TLR-2 is required for maximal MKP-1 induction by S. aureus.
Unlike in our rolipram experiment, increased MKP-1 mRNA expression wasn’t associated with decreased TNFα mRNA expression. This highlights S. aureus’ unique capacity to promote a dysregulated inflammatory response that overpowers host anti-inflammatory mechanisms. Only when MKP-1 is overexpressed by rolipram, is the host anti-inflammatory response able to attenuate S. aureus-induced TNFα expression. An important limitation of the study was the focus on macrophage cell-lines and BMDMs. Additional studies examining rolipram’s efficacy in an in vivo S. aureus sepsis mouse model would better show the effectiveness of MKP-1 induction during systemic infection.
In conclusion, the high global burden of sepsis is of increasing concern. By understanding the body’s anti-inflammatory mechanisms, we could develop better strategies for treating sepsis. This has never been more pertinent than today as antibiotic-resistant bacteria continue to represent a major obstacle to healthcare systems. The data presented illustrates that MKP-1 is inducted in a TLR-2 dependent fashion and functions to negatively regulate S. aureus-induced inflammatory cytokine production in BMDMs. This insight provides additional avenues of study as well as potential therapeutic targets for sepsis treatment.
We’ve shown that rolipram-induced MKP-1 expression is associated with decreased TNFα mRNA expression. This provides further evidence that medications that upregulate MKP-1, such as corticosteroids, are potent inflammatory mediators [13]. While MKP-1 inducers show promise, current therapeutics are characterized by systemic side effects [21]. Corticosteroids are potent anti-inflammatory medications, but they are associated with metabolic dysfunction (e.g., hyperglycemia, hyperlipidemia) [22]. Rolipram is not associated with metabolic dysfunction, however, it carries a significant risk of nausea and headache [21]. Newer PDE-4 inhibitors (e.g., Roflumilast, Apremilast) are associated with less side effects and are currently approved for autoimmune conditions, such as COPD and severe psoriasis. These medications warrant further consideration as possible adjuncts for sepsis treatment considering their ability to increase MKP-1 and improved side effect profile.
Acknowledgements
We thank Dr. Travis R. Taylor for his technical and intellectual advice during the early stages of the research project.
Author Contributions
MB and ZP conceived of the research project. RW, MW, and ZP provided input on experimental design. RW and YL provided bacteria strains and animal resources, respectively. MP conducted the experiments and statistical analysis. MP and JF contributed to writing and editing the manuscript.
Conflicts of Interest
The authors report no conflicts of interest in regard to the published article.
References
2. Morrissey R, Lee J, Baral N, Tauseef A, Sood A, Mirza M, et al. Demographic and regional trends of sepsis mortality in the United States, 1999-2022. BMC Infect Dis. 2025 Apr 11;25(1):504.
3. Thaden JT, Li Y, Ruffin F, Maskarinec SA, Hill-Rorie JM, Wanda LC, et al. Increased Costs Associated with Bloodstream Infections Caused by Multidrug-Resistant Gram-Negative Bacteria Are Due Primarily to Patients with Hospital-Acquired Infections. Antimicrob Agents Chemother. 2017 Feb 23;61(3):e01709–16.
4. Mayr FB, Yende S, Angus DC. Epidemiology of severe sepsis. Virulence. 2014 Jan 1;5(1):4–11.
5. Enright MC, Robinson DA, Randle G, Feil EJ, Grundmann H, Spratt BG. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc Natl Acad Sci U S A. 2002 May 28;99(11):7687–92.
6. Lowy FD. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest. 2003 May;111(9):1265–73.
7. Fournier B, Philpott DJ. Recognition of Staphylococcus aureus by the innate immune system. Clin Microbiol Rev. 2005 Jul;18(3):521–40.
8. Mohammad M, Ali A, Nguyen MT, Götz F, Pullerits R, Jin T. Staphylococcus aureus lipoproteins in infectious diseases. Front Microbiol. 2022 Oct 3;13:1006765.
9. Liew FY, Xu D, Brint EK, O'Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005 Jun;5(6):446–58.
10. Wancket LM, Frazier WJ, Liu Y. Mitogen-activated protein kinase phosphatase (MKP)-1 in immunology, physiology, and disease. Life Sci. 2012 Feb 13;90(7–8):237–48.
11. Frazier WJ, Wang X, Wancket LM, Li XA, Meng X, Nelin LD, et al. Increased inflammation, impaired bacterial clearance, and metabolic disruption after gram-negative sepsis in Mkp-1-deficient mice. J Immunol. 2009 Dec 1;183(11):7411–9.
12. Wang X, Liu Y. Regulation of innate immune response by MAP kinase phosphatase-1. Cell Signal. 2007 Jul;19(7):1372–82.
13. Moosavi SM, Prabhala P, Ammit AJ. Role and regulation of MKP-1 in airway inflammation. Respir Res. 2017 Aug 10;18(1):154.
14. Pan Y, Xu C, Pan ZK. MKP-1 negative regulates Staphylococcus aureus induced inflammatory responses in Raw264.7 cells: roles of PKA-MKP-1 pathway and enhanced by rolipram. Sci Rep. 2017 Sep 28;7(1):12366.
15. Korhonen R, Hömmö T, Keränen T, Laavola M, Hämäläinen M, Vuolteenaho K, et al. Attenuation of TNF production and experimentally induced inflammation by PDE4 inhibitor rolipram is mediated by MAPK phosphatase-1. Br J Pharmacol. 2013 Aug;169(7):1525–36.
16. Lee J, Komatsu K, Lee BC, Lim JH, Jono H, Xu H, et al. Phosphodiesterase 4B mediates extracellular signal-regulated kinase-dependent up-regulation of mucin MUC5AC protein by Streptococcus pneumoniae by inhibiting cAMP-protein kinase A-dependent MKP-1 phosphatase pathway. J Biol Chem. 2012 Jun 29;287(27):22799–811.
17. McCullough AC, Seifried M, Zhao X, Haase J, Kabat WJ, Yogev R, et al. Higher incidence of perineal community acquired MRSA infections among toddlers. BMC Pediatr. 2011 Oct 27;11:96.
18. Mukherjee S, Chen LY, Papadimos TJ, Huang S, Zuraw BL, Pan ZK. Lipopolysaccharide-driven Th2 cytokine production in macrophages is regulated by both MyD88 and TRAM. J Biol Chem. 2009 Oct 23;284(43):29391–8.
19. Sahoo BR, Basu M, Swain B, Dikhit MR, Jayasankar P, Samanta M. Elucidation of novel structural scaffold in rohu TLR2 and its binding site analysis with peptidoglycan, lipoteichoic acid and zymosan ligands, and downstream MyD88 adaptor protein. Biomed Res Int. 2013;2013:185282.
20. Talreja J, Bauerfeld C, Wang X, Hafner M, Liu Y, Samavati L. MKP-1 modulates ubiquitination/phosphorylation of TLR signaling. Life Sci Alliance. 2021 Sep 27;4(12):e202101137.
21. Fan T, Wang W, Wang Y, Zeng M, Liu Y, Zhu S, et al. PDE4 inhibitors: potential protective effects in inflammation and vascular diseases. Front Pharmacol. 2024 Jun 10;15:1407871.
22. Kulkarni S, Durham H, Glover L, Ather O, Phillips V, Nemes S, et al. Metabolic adverse events associated with systemic corticosteroid therapy-a systematic review and meta-analysis. BMJ Open. 2022 Dec 22;12(12):e061476.