Abstract
Obstructive sleep apnea (OSA), the most common sleep disorder, affects nearly 1 billion people worldwide. Moreover, because most cases of OSA remain undiagnosed and untreated, affected individuals are at risk of serious complications, including ischemic stroke, cardiovascular disease, and cognitive decline. Although OSA is a well-established risk factor for cerebrovascular disease, particularly ischemic stroke, whether it might be an independent risk factor for the development, expansion, and rupture of cerebral aneurysms remains unclear. Whereas the pathology of a dissecting cerebral aneurysm differs from that of true cerebral aneurysm, also commonly referred to as saccular or berry aneurysm, the vascular damage caused by OSA is believed to contribute to weakening of the blood vessel walls, thereby increasing the risk of expansion and rupture of both types of aneurysms.
Here, we describe the case of a 57-year-old woman with class III obesity, a history of snoring, witnessed apnea during sleep, fatigue, and excessive daytime sleepiness for several years prior, who presented to the emergency department with diffuse subarachnoid hemorrhage (SAH) secondary to a ruptured dissecting aneurysm of the vertebral artery. The patient underwent endovascular embolization of this dissecting aneurysm, with subsequent significant clinical neurological improvement. An outpatient work-up revealed severe OSA, which was treated with continuous positive airway pressure (CPAP) therapy with significant improvement in her symptoms.
Keywords
Obstructive sleep apnea, Cerebral aneurysm, Subarachnoid hemorrhage, Stroke, Cerebrovascular disease, Intracranial hemorrhage
Abbreviations
OSA: Obstructive Sleep Apnea; SAH: Subarachnoid Hemorrhage; CPAP: Continuous Positive Airway Pressure; AHI: Apnea-Hypopnea Index; HTN: Hypertension; ECG: Electrocardiogram; CT: Computed Tomography; PSG: Polysomnography; HSAT: Home Sleep Apnea Test; MRI: Magnetic Resonance Imaging; MRA: Magnetic Resonance Angiography; MVP: Micro Vascular Plug.
Introduction
The estimated prevalence of unruptured cerebral aneurysms, which varies among regions and populations, is approximately 2–3% of the adult population, or roughly 1 in 50 people; moreover, an estimated 6.8 million people have an unruptured cerebral aneurysm in the United Sates [1]. The annual rate of rupture of cerebral aneurysm with associated intracranial hemorrhage is approximately 6–10 people per 100,000 people; whereases approximately 30,000 people experience cerebral aneurysm rupture each year in the United States, the rates are higher in Japan and Finland [2]. Approximately 15% of people with a ruptured cerebral aneurysm die before reaching the hospital. Ruptured cerebral aneurysms are fatal in half of all affected individuals; among those who survive, approximately 66% experience permanent neurological deficits [3]. Nearly 500,000 deaths due to cerebral aneurysm rupture occur worldwide each year [4]. Ruptured cerebral aneurysms account for 5–15% of all new strokes [5]. Several identified modifiable risk factors promote cerebral aneurysm formation, growth, and rupture, among which the most common factors are hypertension, cigarette smoking, and heavy alcohol consumption [6]. Although obstructive sleep apnea (OSA) might play a role as a risk factor for the development, expansion, and rupture of cerebral aneurysms, this association has not been demonstrated or fully studied [7]. OSA is strongly linked to cardiac and cerebrovascular diseases, including myocardial infarction, hypertension, ischemic stroke, atrial fibrillation, pulmonary hypertension, carotid stenosis, abdominal aortic aneurysm, and aortic dissection, primarily on the bases of underlying hypertension and atherosclerosis associated with untreated OSA [8]. OSA is believed to be associated with an elevated independent risk of aneurysmal subarachnoid hemorrhage (SAH), although this possibility has not been definitively demonstrated [9]. In one study, the prevalence of intracranial aneurysm in people with OSA was 12.1%, a value significantly higher than observed in people without OSA [10]. Additionally, the incidence of aneurysmal SAH was significantly higher in patients with than without OSA [11]. Moreover, patients with intracranial aneurysms and OSA had larger cerebral aneurysms than patients without OSA [12]. The risk of aneurysmal SAH is significantly higher in patients with a diagnosis of OSA than in patients with other sleep disordered breathing [13]. Given the association between OSA and ruptured cerebral aneurysm, early detection and treatment of OSA are crucial to potentially prevent aneurysm rupture and improve outcomes. Whereas most studies have focused on saccular (true) cerebral aneurysms, the underlying mechanisms of damaging cerebral blood vessels due to OSA that contribute to expansion and rupture of these true aneurysms might potentially be the same as those influencing dissecting cerebral aneurysm or pseudoaneurysm [14]. Herein, we present the case of a patient with SAH secondary to dissecting aneurysm, who had symptoms of OSA for several years prior to her presentation and was eventually diagnosed and treated for OSA.
Case Report
A 57-year- old woman with class III obesity presented to the emergency department with headache and confusion. Her past medical history included anxiety, endometrial carcinoma, status post hysterectomy, gastroesophageal reflux disease, presumed ocular histoplasmosis (POH) of the right eye with partial right vision loss, and a recent laparoscopic cholecystectomy. She had no history of smoking or alcohol use. Her home medications included vitamin D and B, escitalopram, esomeprazole, buspirone, and hydrocodone-acetaminophen as needed for pain. She was reported to have changes in her mental status starting an hour prior to evaluation. Her initial vital signs included blood pressure of 156/102 mmHg, 21 respiration per minute, pulse of 136 per minute, temperature 98.8°F (37.1°C), BMI of 45.93 kg/m2, and SpO2 of 97% on room air. An ECG showed normal sinus rhythm with diffuse T-wave inversion in the inferior and anterolateral leads, and prolonged QT/QTC intervals 510/602ms (Figure 1).
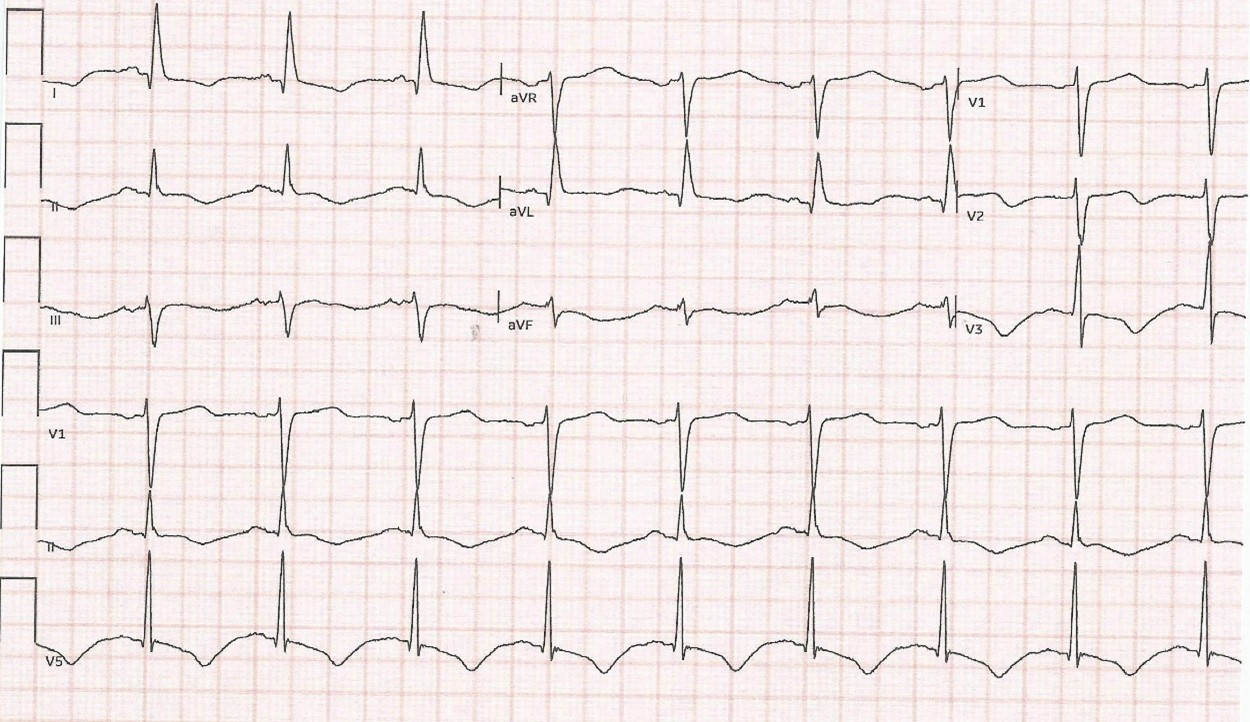
Figure 1. ECG showing diffuse T wave inversion in the inferior and anterolateral leads, with prolonged QT/QTc intervals.
On examination in the emergency department, no external signs of trauma were observed. She was alert but lethargic, and was able to follow commands with no focal neurological deficits. Her initial NIH Stroke Scale score was 0, and her Glasgow coma scale score was 15. A heart examination demonstrated regular rate and rhythm, and a chest examination revealed bilateral rhonchi. Her laboratory data yielded notable findings for glucose (239 mg/dL), Na (137 mEq/L ), K (3.3 mEq/L ), CO2 (17 mEq/L ), anion gap (21), BUN (14 mg/dL), Creatinine (0.9 mg/dL), Troponin (418 ng/L; reference normal range ≤14 ng/L), hemoglobin (13.9 g/dL), hematocrit (43.0%), platelets (360 K/uL), WBC count (17.9 K/uL with neutrophils 44.8%, Lymphocytes 45.3%, monocytes 6%, Eosinophils 2.6%, basophils 0.6% and immature neutrophils 0.7%). Arterial blood gases: pH of 7.25, PCO2 of 51 mm Hg, PO2 of 90 mm Hg, and blood carbon monoxide of 2.1% (reference range 0-5%). A chest X-ray showed bilateral diffuse patchy infiltrates predominantly in the mid to lower lung zones. A CT scan of the head without contrast revealed multicompartment hemorrhages, including diffuse SAHs predominantly within the basal cisterns including the bilateral perimesencephalic, prepontine, quadrigeminal, and suprasellar cisterns, as well as small SAH foci within the bilateral sylvian cisterns, which were associated with fourth ventricle intraventricular hemorrhage and small bilateral subdural hematomas along the tentorium (Figure 2).
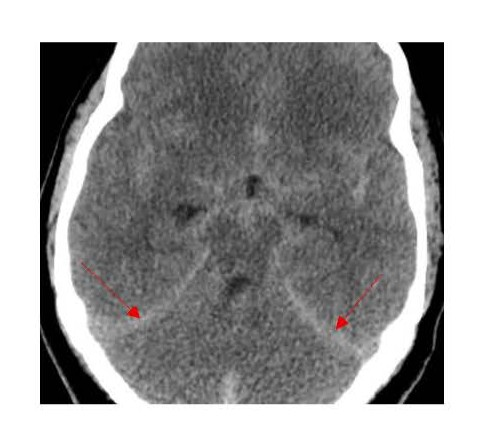
Figure 2. Head CT showing subarachnoid hemorrhage and small tentorial subdural hematomas.
The patient became hypoxic and tachypneic, and showed pink frothy sputum and declining mental status, therefore intubation and mechanical ventilation were required. After blood and urine cultures were obtained, broad-spectrum antibiotics were started because of concerns regarding aspiration pneumonia and sepsis. CT angiography of the head yielded findings concerning for right vertebral artery dissection. She was transferred to our institution for neurosurgery, interventional neurology management, and neurocritical care. On arrival, she received another CT head without contrast, which showed extensive SAH centered at the basal cisterns and surrounding the brainstem, with subdural hematomas along the tentorium and posterior falx, and intraventricular hemorrhage within the occipital horns and the third and fourth ventricles (Figures 3 and 4).
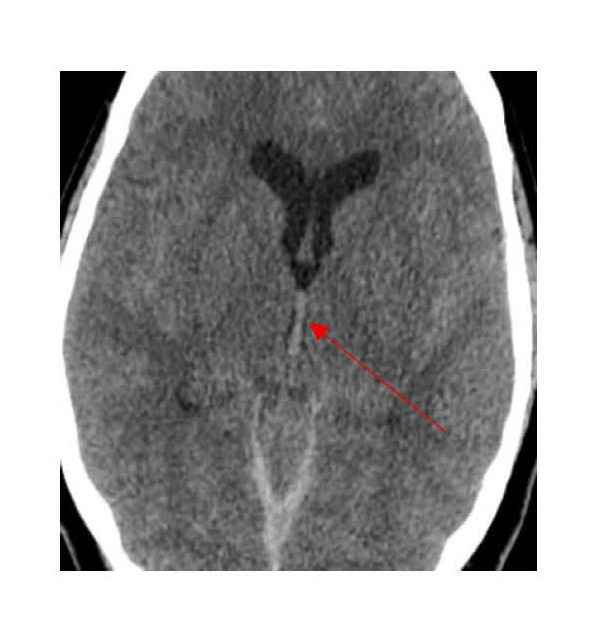
Figure 3. Head CT showing blood in the third ventricle.
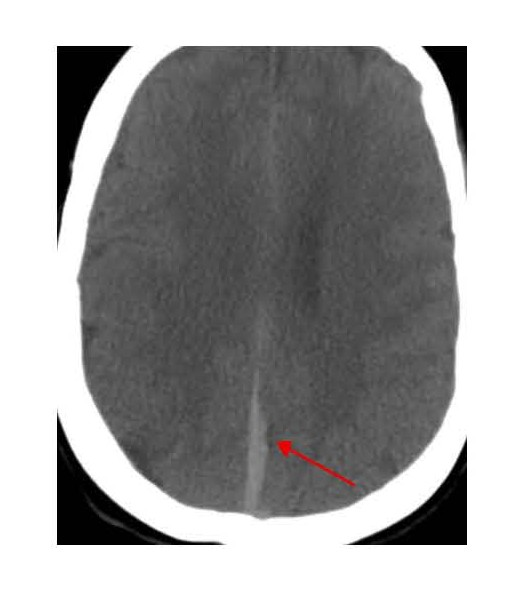
Figure 4. Head CT showing subdural blood along the falx cerebri.
The patient underwent cerebral angiogram demonstrating a dilation of the right vertebral artery in the distal V4 segment just before and proximal to the vertebrobasilar confluence and centered in the region of more prominent hemorrhage on initial scans findings consistent with a ruptured dissecting aneurysm (Figure 5).
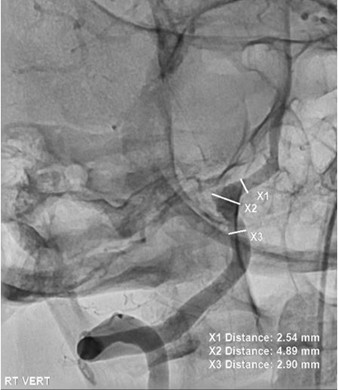
Figure 5. Cerebral angiogram demonstrating a dissecting aneurysm in the right vertebral artery V4 segment.
Given the posterior predominant SAH on CT imaging, we believed that this dissecting aneurysm was the source of the patient’s hemorrhages. The rest of the intracranial circulation was normal with no evidence of other aneurysm or arteriovenous malformation. She underwent endovascular embolization of the right vertebral artery dissecting aneurysm which required vessel sacrifice with coils and a vascular microplug (Figure 6).
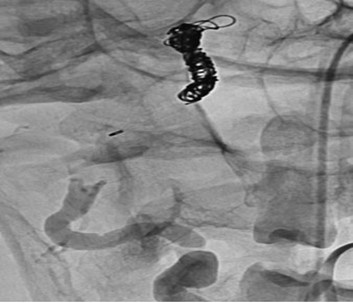
Figure 6. Cerebral angiogram showing successful embolization with coils and MVP of the right vertebral artery dissecting aneurysm requiring vessel sacrifice.
Follow-up angiography demonstrated good occlusion of the dissecting aneurysm and right vertebral artery with no flow in this artery, and continued filling of the posterior circulation through the contralateral vertebral artery. The procedure was successful, and her care continued in the neurocritical care unit. She improved clinically, and was subsequently weaned from mechanical ventilation and successfully extubated in the 24 hours after the procedure, then placed on oxygen with a nasal cannula at 4 L/min. Her ECG continued to show T wave inversion and QT/QTc prolongation while she was not taking escitalopram. Cardiac evaluation with echocardiography demonstrated a mildly dilated left ventricle with normal wall thickness; normal ventricular mass; and preserved motion of the basal anterior, anterolateral, inferior, and inferoseptal segments of the left ventricle. The remaining segments were hypokinetic, and the apex appeared severely hypokinetic, with moderately reduced systolic function and an estimated ejection fraction of 30–35% with grade I diastolic dysfunction. These findings suggested stress-induced cardiomyopathy (Takotsubo cardiomyopathy) related to SAH.
On day 12 of hospitalization, she underwent left heart catheterization and coronary angiography with left ventriculography, which showed patent coronaries, normal left ventricle dimensions, no regional wall motion abnormality, and a normal left ventricular ejection fraction (estimated to be 70%). The diffuse T wave inversion and QT/QTc intervals prolongation seen on her initial ECG were believed to be associated with SAH rather than coronary ischemia. Her initial acute hypoxic respiratory failure was believed to be related to cardiogenic pulmonary edema secondary to underlying Takotsubo cardiomyopathy associated with SAH and/or neurogenic pulmonary edema due to SAH. Notably, at the time of hospitalization, she was reportedly snoring during her sleep.
After she had stabilized, physical and occupational therapy were started. A swallowing evaluation by videofluoroscopy demonstrated significant dysphagia requiring percutaneous endoscopic gastrostomy tube (PEG) placement for feeding.
The patient was discharged to inpatient rehabilitation for ongoing physical and occupational therapy, as well as skilled speech therapy. Eventually, the percutaneous endoscopic gastrostomy tube was removed after her dysphagia resolved. She was discharged home and made a remarkable recovery: she was able to return to work, drive, and participate in all her usual activities of daily life with full functional recovery. Her Modified Rankin Scale Score at 1 year follow up was 0. MRA of the brain at 3 months and one year demonstrated occlusion of the dissecting aneurysm and right vertebral artery with good flow in the posterior circulation via the left vertebral artery.
The patient was evaluated for sleep disordered breathing. She reported having snoring, excessive daytime sleepiness, fatigue, witnessed apnea, and waking up gasping for air for several years before her current illness. Her Epworth scale score was 16. She underwent polysomnography (PSG), which demonstrated an AHI of 49.1, finding consistent with severe OSA with nocturnal hypoxemia, a lowest nocturnal oxygen saturation 71%, and no central sleep apnea. A CPAP titration study demonstrated that a CPAP pressure of 14 cmH2O was needed to control her underlying OSA. She was prescribed a CPAP of 14 cmH2O with a full-face mask in addition to a weight reduction program. In a follow-up visit, she reported a significant improvement in her OSA symptoms, and the CPAP compliance report indicated resolution of her OSA on CPAP therapy and excellent CPAP adherence.
Discussion
Non-traumatic SAH is associated with a high mortality rate (as high as 45%–50% in the first 30 days) and high morbidity. Survivors commonly have high disability rates, and impaired functional capacity and quality of life. Most non-traumatic SAH cases are caused by rupture of intracranial aneurysms. Risk factors for rupture of cerebral aneurysm include aneurysm size [15,16], posterior circulation location, and aneurysm shape: irregular aneurysms with daughter sacs are correlated with high risk of rupture, as well as smoking history and uncontrolled hypertension.
Known modifiable risk factors contributing to the formation of cerebral aneurysm include hypertension and cigarette smoking, whereas non-modifiable risk factors include family history of cerebral aneurysm, older age, and female sex. Genetic predispositions to cerebral aneurysm include Ehlers Danlos syndrome, autosomal dominant polycystic kidney disease, Marfan syndrome, hereditary hemorrhagic telangiectasia, fibromuscular dysplasia, alpha 1 antitrypsin deficiency, multiple endocrine neoplasia type I (MEN1), pseudoxanthoma elasticus, Moya-moya disease, sickle cell disease, bicuspid aortic valve, coarctation of the aorta, tuberos sclerosis, neurofibromatosis type 1, pheochromocytoma, and Klinefelter syndrome [17]. However, cerebral aneurysms secondary to these genetic disorders account for less than 1% of all cerebral aneurysms in the general population [18]. Other risk factors remain unrecognized.
Although OSA has not been demonstrated to be a risk factor for cerebral aneurysm growth and rupture, it has a potential role in the pathogenesis of the blood vessel wall disease. OSA has been identified as an independent risk factor for cerebrovascular disease (particularly ischemic stroke), myocardial infarction, hypertension, atrial fibrillation, carotid stenosis, pulmonary hypertension, abdominal aortic aneurysm, and aortic dissection. Additionally, the risk of stroke in people with OSA increases with the severity of the underlying OSA [19–21]. Moderate to severe OSA has been independently associated with elevated risk of cerebral microbleeds (CMB) in middle-aged and older adults, which in turn is associated with elevated risk of developing stroke, accelerated cognitive decline, and dementia [22]. Several studies have found a strong association between OSA and the development and progression of abdominal and thoracic aneurysms [23–25]. However, OSA has remained an underrecognized risk factor for cerebral aneurysm and SAH, and its association remains inconclusive. If this link is demonstrated and confirmed, OSA might potentially be a treatable and modifiable risk factor that could prevent aneurysm rupture. Preventive measures to limit cerebral aneurysms formation, growth, and rupture are important to reduce catastrophic complications such as SAH and thereby improve outcomes by identifying modifiable risk factors early in the disease course.
People with OSA have a higher incidence of aneurysmal SAH than the general population or individuals with other sleep disorders. One study has indicated an incidence rate of 48.2% per 100,000 person-years among patients with OSA versus a general rate of approximately 9–14% per 100,000 person-years in patients without OSA [26]. Moreover, patients with OSA are more likely to have significantly larger ruptured cerebral aneurysms than patients without OSA who experience cerebral aneurysms rupture [27]. In addition, patients with OSA have been reported to have poorer outcomes if they develop SAH due to cerebral aneurysmal rupture than patients with aneurysmal SAH but without OSA [28]. Furthermore, cerebral vasospasm after cerebral aneurysm rupture is more frequently seen in patients with OSA than in patients without OSA.
The effects of OSA on cerebral aneurysm development and expansion might be partly mediated by hypertension [29], thus placing vascular shear stress on vessel walls [30], in addition to increased sympathetic activity due to intermittent nocturnal hypoxia [31]. Other mechanisms potentially responsible for the development of cerebral aneurysms in people with OSA might include oxidative stress and reduced endothelial regeneration [32]. Additionally, OSA is associated with systemic and local inflammation with elevated plasma concentrations of several inflammatory markers including cytokines [33], interleukin-6, tumor necrosis factor alpha (TNF-alpha), acute phase protein, endothelial adhesion molecules, matrix metalloproteinase, C-reactive protein, and vascular endothelial growth factor (VEGF), because of intermittent hypoxia during apneic episodes [34]. Moreover, diminished nitric oxide availability can lead to vascular endothelial injury [35,36], endothelial apoptosis [37], and endothelial dysfunction with vascular remodeling, all of which can contribute to blood vessel wall damage, accelerate atherosclerosis, and lead to aneurysm formation, growth and rupture. These mechanisms of vascular damage, in addition to the intrathoracic pressure changes during sleep seen in people with OSA, have important roles in promoting thoracic and abdominal aortic aneurysm expansion and rupture [38].
Dissecting cerebral aneurysms and pseudoaneurysms are different entities that could arise in relationship with trauma, post-surgical injury to the arteries, or dissection, or may be secondary to infection, neoplasia, or radiation therapy. Unlike saccular aneurysms, pseudoaneurysms lack a complete arterial wall with all layers and therefore might be unstable and more vulnerable to the damaging effect of OSA. For example, fluctuations and intense blood pressure surges during apnea events might increase mechanical stress and exert greater force on already weakened areas and fragile blood vessel walls. Consequently, pseudoaneurysms may expand and eventually rupture thus leading to intracranial hemorrhage [39]. The endothelial dysfunction and oxidative stress associated with OSA can damage the endothelium, the inner lining of blood vessels, and potentially contribute to pseudoaneurysm growth. Theoretically, a dissecting aneurysm or a pseudoaneurysm might have greater risk of expansion and rupture from the effect of OSA than a saccular aneurysm.
Although OSA has been demonstrated to be an independent risk factor for ischemic stroke, no current studies have directly linked OSA to the development and rupture of cerebral aneurysms, arterial dissection and dissecting pseudoaneurysms. Further research is needed to fully understand the association between OSA and these conditions. However, given the potential associations, evaluating patients with a diagnosed cerebral aneurysm or non-traumatic SAH for possible OSA who have symptoms of OSA is highly recommended.
Several screening tools for OSA, such as the Epworth Sleepiness Scale (ESS) [40], Stanford Sleepiness Scale (SSS) [41], STOP-bang criteria [42,43], and Berline questionnaires [44], have been used and validated as subjective measures of sleepiness, and have varying sensitivity and specificity depending on the cutoff score. Although these screening tools are useful for risk stratification, they should always be used in conjunction with a comprehensive clinical assessment and a diagnostic sleep test [45]. PSG, the gold standard for OSA diagnosis, is performed in a sleep laboratory. A home sleep apnea test (HSAT) is an acceptable alternative for patients without significant comorbidities; this method is appropriate for patients with a high pretest probability of OSA and achieves reasonable sensitivity and specificity and an overall accuracy of 83–91% for moderate and severe OSA. However, HSAT have lower accuracy than PSG. CPAP is highly effective and remains the gold standard of care for most patients with OSA, yet low adherence and patient intolerance pose substantial challenges [46]. Mandibular advancement device (MAD) and upper airway stimulation (UAS), also known as hypoglossal nerve stimulation (HSN), are alternatives for patients with OSA who cannot tolerate CPAP. Patients are eligible for UAS if their BMI is < 35–40 kg/m2, their AHI is >15, and CPAP is either intolerable or unable to control their OSA. Custom-made mandibular advancement devices are mostly efficacious in mild to moderate OSA and have higher patient adherence rate than CPAP. CPAP therapy has been shown to reverse vascular endothelial dysfunction, inflammation, and enhance endothelial repair capacity. Treating patients with OSA with CPAP may potentially lower the risk of stroke in high-risk patients [47].
Conclusion
OSA is an established independent risk factor for cerebrovascular disease, particularly ischemic stroke [48]. Although the literature suggests an association between OSA and mechanisms that could lead to the development, expansion, and rupture of cerebral aneurysms, additional randomized prospective-controlled studies are needed to establish whether an association might exist between OSA and cerebral aneurysms, identify the mechanisms involved, and determine whether OSA might be considered a modifiable risk factor for cerebral aneurysm formation, growth and rupture, to support guidelines regarding preventive measures. Currently, no formal recommendation or guidance is available for screening patients with cerebral aneurysms for OSA; however, screening for OSA in patients with diagnosed intracranial aneurysms and symptoms suggestive of OSA might identify modifiable risk factors for cerebrovascular disease.
References
2. Wiebers DO, Whisnant JP, Huston J 3rd, Meissner I, Brown RD Jr, Piepgras DG, et al. Unruptured intracranial aneurysms: natural history, clinical outcome, and risks of surgical and endovascular treatment. Lancet. 2003 Jul 12;362(9378):103–10.
3. Sacco S, Marini C, Toni D, Olivieri L, Carolei A. Incidence and 10-year survival of intracerebral hemorrhage in a population-based registry. Stroke. 2009 Feb;40(2):394–9.
4. Rost NS, Smith EE, Chang Y, Snider RW, Chanderraj R, Schwab K, et al. Prediction of functional outcome in patients with primary intracerebral hemorrhage: the FUNC score. Stroke. 2008 Aug;39(8):2304–9.
5. Manhas A, Nimjee SM, Agrawal A, Zhang J, Diaz O, Zomorodi AR, et al. Comprehensive Overview of Contemporary Management Strategies for Cerebral Aneurysms. World Neurosurg. 2015 Oct;84(4):1147–60.
6. Bacigaluppi S, Piccinelli M, Antiga L, Veneziani A, Passerini T, Rampini P, et al. Factors affecting formation and rupture of intracranial saccular aneurysms. Neurosurg Rev. 2014 Jan;37(1):1–14.
7. Zaremba S, Güresir E. Is there a causal relationship between obstructive sleep apnea and the pathophysiology of intracranial aneurysm?. Somnologie. 2019 Mar;23(1):29–35.
8. Peker Y, Hedner J, Norum J, Kraiczi H, Carlson J. Increased incidence of cardiovascular disease in middle-aged men with obstructive sleep apnea: a 7-year follow-up. Am J Respir Crit Care Med. 2002 Jul 15;166(2):159–65.
9. Inamasu J, Akiyama T, Akaji K, Inaba M, Nishimoto M, Kojima A, et al. Aneurysmal subarachnoid hemorrhage occurring during sleep: Clinical characteristics and risk factors. J Stroke Cerebrovasc Dis. 2024 Apr;33(4):107591.
10. Jung TY, Lee E, Park M, Lee JY, Hong YS, Cho J, et al. Obstructive Sleep Apnea and Its Influence on Intracranial Aneurysm. J Clin Med. 2023 Dec 27;13(1):144.
11. Alaqeel AM, Almasri SH, Alotaibi NM, Al-Yamany MA, Bahammam AS, Mohammad YM, et al. Prevalence of symptoms and risk of sleep apnea in patients with ruptured cerebral aneurysm. Neurosciences (Riyadh). 2013 Jul;18(3):248–51.
12. Zaremba S, Albus L, Hadjiathanasiou A, Vatter H, Wüllner U, Güresir E. Aneurysm size and blood pressure severity in patients with intracranial aneurysms and sleep apnea. J Clin Sleep Med. 2022 Jun 1;18(6):1539–45.
13. Chernyshev OY, Bir SC, Maiti TK, Patra DP, Sun H, Guthikonda B, et al. The Relationship Between Obstructive Sleep Apnea and Ruptured Intracranial Aneurysms. J Clin Sleep Med. 2019 Dec 15;15(12):1839–48.
14. Danishara MA, Al-Fauzi A, Hidayati HB. Cerebral pseudoaneurysms: a review of the current literature. WJARR. 2014;24(3):1714–18.
15. Shojima M, Morita A, Nakatomi H, Tominari S. Size is the Most Important Predictor of Aneurysm Rupture Among Multiple Cerebral Aneurysms: Post Hoc Subgroup Analysis of Unruptured Cerebral Aneurysm Study Japan. Neurosurgery. 2018 Jun 1;82(6):864–9.
16. Wermer MJ, van der Schaaf IC, Algra A, Rinkel GJ. Risk of rupture of unruptured intracranial aneurysms in relation to patient and aneurysm characteristics: an updated meta-analysis. Stroke. 2007 Apr;38(4):1404–10.
17. Lindbohm JV, Kaprio J, Jousilahti P, Salomaa V, Korja M. Sex, Smoking, and Risk for Subarachnoid Hemorrhage. Stroke. 2016 Aug;47(8):1975–81.
18. Toader C, Eva L, Bratu BG, Covache-Busuioc RA, Costin HP, Dumitrascu DI, et al. Intracranial Aneurysms and Genetics: An Extensive Overview of Genomic Variations, Underlying Molecular Dynamics, Inflammatory Indicators, and Forward-Looking Insights. Brain Sci. 2023 Oct 12;13(10):1454.
19. Shahar E, Whitney CW, Redline S, Lee ET, Newman AB, Nieto FJ, et al. Sleep-disordered breathing and cardiovascular disease: cross-sectional results of the Sleep Heart Health Study. Am J Respir Crit Care Med. 2001 Jan;163(1):19–25.
20. Yeghiazarians Y, Jneid H, Tietjens JR, Redline S, Brown DL, El-Sherif N, et al. Obstructive Sleep Apnea and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation. 2021 Jul 20;144(3):e56–e67.
21. Gaisl T, Bratton DJ, Kohler M. The impact of obstructive sleep apnoea on the aorta. Eur Respir J. 2015 Jul 31;46(2):532–44.
22. Siddiquee AT, Hwang YH, Kim S, Lee SK, Lee MH, Kim HJ, et al. Obstructive Sleep Apnea and Cerebral Microbleeds in Middle-Aged and Older Adults. JAMA Netw Open. 2025 Oct 1;8(10):e2539874.
23. Mason RH, Ruegg G, Perkins J, Hardinge M, Amann-Vesti B, Senn O, et al. Obstructive sleep apnea in patients with abdominal aortic aneurysms: highly prevalent and associated with aneurysm expansion. Am J Respir Crit Care Med. 2011 Mar 1;183(5):668–74.
24. Saruhara H, Takata Y, Usui Y, Shiina K, Hashimura Y, Kato K, et al. Obstructive sleep apnea as a potential risk factor for aortic disease. Heart Vessels. 2012 Mar;27(2):166–73.
25. Zhai T, Liu B, Zhang J, Wu Y. Impact of obstructive sleep apnea on aortic disease occurrence: A meta-analysis. Heliyon. 2022 Aug 1;8(8):e10049.
26. Zaremba S, Albus L, Schuss P, Vatter H, Klockgether T, Güresir E. Increased risk for subarachnoid hemorrhage in patients with sleep apnea. J Neurol. 2019 Jun;266(6):1351–7.
27. Geer JH, Falcone GJ, Vanent KN, Leasure AC, Woo D, Molano JR, et al. Obstructive Sleep Apnea as a Risk Factor for Intracerebral Hemorrhage. Stroke. 2021 May;52(5):1835–8.
28. Bir SC, Nanda A, Cuellar H, Sun H, Guthikonda B, Liendo C, et al. Coexistence of obstructive sleep apnea worsens the overall outcome of intracranial aneurysm: a pioneer study. J Neurosurg. 2018 Mar;128(3):735–46.
29. Tada Y, Wada K, Shimada K, Makino H, Liang EI, Murakami S, et al. Roles of hypertension in the rupture of intracranial aneurysms. Stroke. 2014 Feb;45(2):579–86.
30. Pedrosa RP, Drager LF, Gonzaga CC, Sousa MG, de Paula LK, Amaro AC, et al. Obstructive sleep apnea: the most common secondary cause of hypertension associated with resistant hypertension. Hypertension. 2011 Nov;58(5):811–7.
31. Garvey JF, Taylor CT, McNicholas WT. Cardiovascular disease in obstructive sleep apnoea syndrome: the role of intermittent hypoxia and inflammation. Eur Respir J. 2009 May;33(5):1195–205.
32. Jelic S, Padeletti M, Kawut SM, Higgins C, Canfield SM, Onat D, et al. Inflammation, oxidative stress, and repair capacity of the vascular endothelium in obstructive sleep apnea. Circulation. 2008 Apr 29;117(17):2270–8.
33. Jelic S, Le Jemtel TH. Inflammation, oxidative stress, and the vascular endothelium in obstructive sleep apnea. Trends Cardiovasc Med. 2008 Oct;18(7):253–60.
34. Imagawa S, Yamaguchi Y, Higuchi M, Neichi T, Hasegawa Y, Mukai HY, et al. Levels of vascular endothelial growth factor are elevated in patients with obstructive sleep apnea--hypopnea syndrome. Blood. 2001 Aug 15;98(4):1255–7.
35. Nadeem R, Molnar J, Madbouly EM, Nida M, Aggarwal S, Sajid H, et al. Serum inflammatory markers in obstructive sleep apnea: a meta-analysis. J Clin Sleep Med. 2013 Oct 15;9(10):1003–12.
36. Dyugovskaya L, Lavie P, Lavie L. Increased adhesion molecules expression and production of reactive oxygen species in leukocytes of sleep apnea patients. Am J Respir Crit Care Med. 2002 Apr 1;165(7):934–9.
37. El Solh AA, Akinnusi ME, Baddoura FH, Mankowski CR. Endothelial cell apoptosis in obstructive sleep apnea: a link to endothelial dysfunction. Am J Respir Crit Care Med. 2007 Jun 1;175(11):1186–91.
38. Ryan S, Taylor CT, McNicholas WT. Systemic inflammation: a key factor in the pathogenesis of cardiovascular complications in obstructive sleep apnoea syndrome? Thorax. 2009 Jul;64(7):631–6.
39. Drager LF, Polotsky VY, Lorenzi-Filho G. Obstructive sleep apnea: an emerging risk factor for atherosclerosis. Chest. 2011 Aug;140(2):534–42.
40. Banhiran W, Assanasen P, Nopmaneejumruslers C, Metheetrairut C. Epworth sleepiness scale in obstructive sleep disordered breathing: the reliability and validity of the Thai version. Sleep Breath. 2011 Sep;15(3):571–7.
41. Hoddes E, Dement W, Zarcone V. The development and use of the Stanford sleepiness scale (SSS). Psychophysiology. 1972;9:150.
42. Shahid A, Wilkinson K, Marcu S, Shapiro CM, et al. STOP, THAT and one hundred other sleep scales. Springer Science+ Business Media, LLC 2012;369–70.
43. Pivetta B, Chen L, Nagappa M, Saripella A, Waseem R, Englesakis M, et al. Use and Performance of the STOP-Bang Questionnaire for Obstructive Sleep Apnea Screening Across Geographic Regions: A Systematic Review and Meta-Analysis. JAMA Netw Open. 2021 Mar 1;4(3):e211009.
44. Netzer NC, Stoohs RA, Netzer CM, Clark K, Strohl KP. Using the Berlin Questionnaire to identify patients at risk for the sleep apnea syndrome. Ann Intern Med. 1999 Oct 5;131(7):485–91.
45. Vanagas T, Lipskytė D, Tamošiūnaitė J, Petrikonis K, Pajėdienė E. Can We Reduce the Diagnostic Burden of Sleep Disorders? A Single-Centre Study of Subjective and Objective Sleep-Related Diagnostic Parameters. Medicina (Kaunas). 2025 Apr 23;61(5):780.
46. Weaver TE, Grunstein RR. Adherence to continuous positive airway pressure therapy: the challenge to effective treatment. Proc Am Thorac Soc. 2008 Feb 15;5(2):173–8.
47. Davis AP, Billings ME, Longstreth WT Jr, Khot SP. Early diagnosis and treatment of obstructive sleep apnea after stroke: Are we neglecting a modifiable stroke risk factor? Neurol Clin Pract. 2013 Jun;3(3):192–201.
48. Bassetti CL, Milanova M, Gugger M. Sleep-disordered breathing and acute ischemic stroke: diagnosis, risk factors, treatment, evolution, and long-term clinical outcome. Stroke. 2006 Apr;37(4):967–72.