Abstract
Significant progress has been made in the advancement of immune system modulation for cancer treatment in recent years. In particular, immune checkpoint inhibitors and chimeric antigen receptor (CAR) T-cell therapy have demonstrated remarkable clinical benefit in relapsed/refractory cancers. However, our understanding of the immuno-oncologic landscape in pediatric solid tumors remains limited and is a barrier to continued progress. We examined the immunohistochemical expression of checkpoint receptors PD-1, TIM-3, LAG-3 and their respective ligands in various pediatric cancers at diagnosis and found high expression of TIM-3/Galectin-9 in the infiltrating cells of Ewing sarcoma. Location of checkpoint receptor/ligand expressions is important, as some staining patterns were only seen along tumor borders. Finally, peripheral T cell function varied significantly among different tumors supporting a complex relationship between the tumor microenvironment and the global immune system.
Abbreviations
PD-1: Programmed Death receptor; PD-L1: Programmed Death receptor Ligand 1; LAG-3: Lymphocyte Activating Gene 3; TIM-3: T cell Immunoglobulin and Mucin domain containing 3; CTLA-4: Cytotoxic T-Lymphocyte-Associated protein 4; FDA: Food and Drug Administration; MHC: Major Histocompatibility Complex; NHL: Non-Hodgkin Lymphoma; DLBCL: Diffuse Large B Cell Lymphoma; PMBCL: Primary Mediastinal B Cell Lymphoma; ANOVA: Analysis of Variance; SR B-ALL: Standard Risk B-cell Acute Lymphoblastic Leukemia; TILs: Tumor Infiltrating Lymphocytes; BTLA: B and T Lymphocyte Attenuator; JAK/ STAT: Janus Activated Kinase/Signal Transducer and Activator of Transcription; TIGIT: T cell Immunoreceptor with Ig and ITIM domains; CAR: Chimeric Antigen Receptor; IFN-γ: Interferon Gamma; AP-1: Activator Protein 1; IRF: Interferon Regulatory Factor; NCR: Negative Checkpoint Receptor
Introduction
The concept of immunotherapy has been traced as far back to third century BC Qin dynasty in China through the purposeful inoculation of Variola minor to prevent smallpox [1]. Harnessing the knowledge gained through the triumphs of vaccination, use of immune-mediated therapies to treat cancer was first pioneered by William B. Coley, a nineteenth century surgeon at the Hospital for Ruptured and Crippled (now the Hospital for Special Surgery), who noticed that concomitant infection with erysipelas seemed to cause the remission of metastatic sarcoma [2].
Over the next few decades, continued understanding in immunobiology and technologic advancements have led to the exciting development of genetically engineered T cells and immune checkpoint inhibitors that is revolutionizing the treatment of childhood cancers [3,4].
Of particular interest are the recent advances using immune checkpoint blockade as a tool to harness the power of the immune system to fight cancer [5]. When T cells are under constant exposure to cancer cells, they become exhausted and are increasingly less apt in clearing the antigen [6]. Exhausted T cells are unique in that they have a progressive and hierarchical loss of effector functions, altered expression of key transcription factors, and specific metabolic derangements. Furthermore, they also express multiple inhibitor receptors such as Programmed death receptor (PD-1), Lymphocyte activating gene 3 (LAG-3), and T cell immunoglobulin and mucin domain containing 3 (Tim-3) [7]. Targeting of these checkpoint receptors within the tumor microenvironment serves as the basis for the development of drugs such as Nivolumab, a PD-1 inhibitor, that can effectively reverse T cell’s dysfunctional state mediated by the PD-1 pathway and therefore reinvigorate immune responses.
In adult cancer studies, expression of checkpoint receptors and their respective ligands have been demonstrated on immunohistochemistry staining across various tumors. Interestingly, the density, location, and cell types that express these receptors and ligands vary between and within tumor types thus contributing to varying clinical responses to checkpoint inhibitors [8-13]. Nonetheless, Ipilimumab (targeting Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)) showed an impressive improvement in overall survival in adult patients with advanced melanoma and became the first FDA approved immune checkpoint inhibitor in 2010 [14]. Since then, hundreds of phase I/II trials are underway to evaluate the efficacy of multiple immune checkpoint inhibitor monotherapy or in combination [13].
Owing to the success of checkpoint inhibitors in adult clinical trials, there are now 2 checkpoint inhibitors Ipilimumab and Pembrolizumab that have been approved by the United States Food and Drug Administration (FDA) for pediatric patients with advanced melanoma and relapsed/refractory classical Hodgkin lymphoma respectively [13,15,16]. Much of the pediatrics studies to date have focused on only Programmed death receptor ligand 1 (PD-L1) and PD-1 expression and results have been mixed. Reported PD-L1 expression in at least 1% of tumor cells varies from 9% to 33%, and most samples were from recurrent or metastatic disease [17-22]. Furthermore, LAG-3 and Tim-3 have both gained substantial interest in adult cancer therapy leading to multiple anti-LAG-3 and anti-Tim-3 clinical trials (NCT01968109, NCT02817633). However, the availability of such clinical trials has not yet progressed to pediatric patients as the preclinical data on the expression of these checkpoint receptors on pediatric tumors remains limited.
As such, there is a great need for better understanding of the onco-immunologic landscape of pediatric tumors in order to provide more targeted therapeutic strategies. In this study, we characterized the expression of PD- 1, LAG-3, Tim-3, and their respective ligands PD-L1, major histocompatibility complex class II (MHC class II), Galectin-9 in children with Ewing sarcoma and osteosarcoma at diagnosis. We also assessed the interplay between tumor and the effects on circulating T cells to determine the global severity of T cell dysfunction. Examination of the immune-oncologic landscape of these tumor types will shed insight upon potential checkpoint inhibitor therapy, as well as peripheral T cell biomarkers for cellular therapy, as the number of checkpoint receptors simultaneously expressed can be reflective of the severity of T cell dysfunction [6,23].
Materials and Methods
Patient selection and clinical protocol
Twenty-five patients with newly diagnosed malignancies were enrolled as part of a clinical trial under an approved Children’s Hospital of Philadelphia Institutional Review Board protocol 12-009915, with written informed consent obtained in accordance with the U.S. common rule (Table 1). Patient age at time of enrollment ranged from 5 years to 22 years of age. Disease groups included Ewing sarcoma (n=17), and osteosarcoma (n=8). Biopsy specimens were obtained at diagnosis (prior to chemotherapy or radiation) via either open biopsy or core-needle biopsy based on surgeon preference. Of all biopsy specimens, 7 of 17 Ewing sarcoma samples and 0 of 10 osteosarcoma samples displayed a definite tumor border as defined by juxtaposition of normal tissue and tumor.
| Disease | Age (years) | Number of patients | Biopsies with tumor border |
|---|---|---|---|
| Ewing sarcoma | 12 ± 5 | 10 | 6 of 10 |
| NHL | 18 ± 4.7 | 8 | 3 of 8 |
| Osteosarcoma | 16 ± 4.6 | 8 | 0 of 10 |
IHC staining
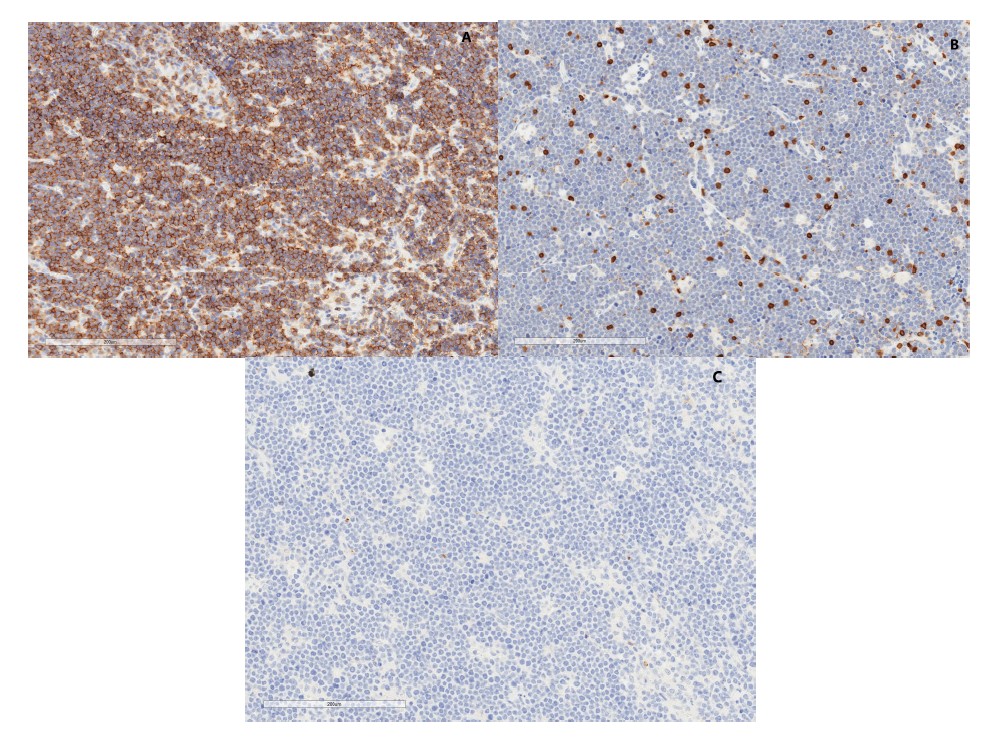
Formalin fixed paraffin embedded tissue sections were stained with antibodies detailed in (Table 2). Slides were stained with a Leica Bond autostainer (Leica Biosystems) using the Refine staining kit (DS9800). The standard protocol F was followed with the exception of the primary antibody incubation time which was extended to 60 minutes. For fluorescent staining (ALK, Galectin 9), slides were processed by hand using standard methods. Briefly, slides were deparaffinized in xylene and rehydrated in a descending series of ethanol. Antigen retrieval was performed using a pressure cooker (Biocare Medical) with antigen unmasking solution (Vector Labs). Blocking was performed in 0.5% SDS for 20 minutes, followed by 2% fetal bovine serum for 20 minutes. Primary antibodies were incubated at room temperature for 60 minutes followed by appropriate Alexa fluor 488 or 594 Secondary antibody incubation (Invitrogen) at a 1:200 dilution for 1 hour at room temperature. Slides were counter stained with DAPI (Sigma) and scanned at 20x magnification using an Aperio CS-O (brightfield) or fluorescent scanner. Results of IHC staining pattern were reviewed with a single pathologist and graded as “negative”, “rare” or “positive” (Figure 1).
| Antibody | Clone | Vendor | Catalog Number | Dilution | Antigen Retrieval | Protocol |
|---|---|---|---|---|---|---|
| CD20 | L26 | Dako | M0755 | 1:1K | E2-20 | Leica Bond Refine |
| CD68 | Kp1 | Dako | M0814 | 1:4K | E2-20 | Leica Bond Refine |
| PD1 | EH33 | Cell Signaling | 43248 | 1:100 | E2-20 | Leica Bond Refine |
| Tim-3 | D5DR | Cell Signaling | 45208 | 1:50 | E2-20 | Leica Bond Refine |
| Lag-3 | 17B4 | LSBio | C18692 | 1:200 | E1-20 | Leica Bond Refine |
| PD-L1 | E13N(R) | Cell Signaling | 13684 | 1:200 | E1-20 | Leica Bond Refine |
| CD3 | n/a | Dako | A0452 | 1:100 | E1-20 | Leica Bond Refine |
| CD56 | VRQ-42 | Cell Marque | 156-R-95 | 1:400 | E2-20 | Leica Bond Refine |
| ALK | D5F3 | Cell Signaling | 3633 | 1:200 | P.C. Citrate | IF |
| Galectin 9 | n/a | R&D Systems | AF2045 | 1:50 | P.C. Citrate | IF |
| MHCII HLA-DR | TAL.1B5 | Dako | M0746 | 1:100 | E1-20 | Leica Bond Refine |
Flow cytometry
Flow cytometry was performed on peripheral blood T cells with the following antibodies: PD1-BV510 (BD Biosciences, #563076), Tim3-BV421 (BD Biosciences, #565562), and LAG3-PerCP-eFluor 710 (BD Biosciences, #3DS223H). Samples were washed twice and flow cytometry was conducted with FACS Verse (BD Biosciences). Due to lack of available samples, only seven peripheral blood samples were analyzed from the osteosarcoma group and 8 peripheral blood samples were analyzed from the Ewing sarcoma group.
Ex vivo T cell expansion and culture
Lymphocytes were harvested from peripheral blood and expanded using CD3/CD28 beads as previously described [24]. After 7 days, beads were magnetically removed and cells were cultured for an additional seven days. Fold expansion was calculated by dividing maximum cell count by the starting cell count.
Statistical analysis
Flow cytometry data were analyzed using FlowJo software (v10.6.1; Tree Star). Subsequent statistical analysis was performed using Prism 8 (Version 8.2.0, Graphpad Software, La Jolla, CA) using analysis of variance (ANOVA) testing for group comparisons. Significant results are denoted by p-value <0.05.
Gene expression analysis
RNA was isolated using the RNeasy mini plus kit (QIAGEN) and hybridized to the nCounter CAR-T characterization panel (human codeset). RCC files containing raw counts for 770 genes provided from NanoString were loaded into nSolver Analysis Software 3.0 and normalized for housekeeping genes and positive controls. Sequence data was analyzed using Morpheus (Broad Institute) to map transcripts and calculate reads per kilobase million (SW780 cDNA expression) or transcripts per million (UM-UC-9).
Results
Negative checkpoint receptors (NCR) and ligands expression pattern in pediatric tumors vary based upon tumor type and location within the tumor
Eighteen biopsy samples were stained with PD-1, PD-L1, TIM-3, Galectin-9, LAG-3, and MHC Class II and underwent pathology review. Tumor infiltrating lymphocytes (TILs) were differentiated against tumor cells based on expression of CD3, CD20, or CD68 on IHC staining. For Ewing sarcoma, 4 of 10 (40%) samples had positive PD-1 staining of tumor cells, and 10 of 10 (100%) samples had positive staining for both TIM-3 and Galectin-9 in infiltrating non-tumor cells (Table 3A). To confirm our findings, 7 additional diagnostic Ewing sarcoma biopsy samples demonstrated 7 of 7 (100%) positive staining for TIM-3, and 5 of 7 (71%) positive staining for Galectin-9 ( Table 3B). This expression pattern is different from osteosarcoma staining as none of the samples had positive PD-1 staining of tumor cells, and only 5 of 8 (62.5%) osteosarcoma samples stained positively for both TIM-3 and Galectin-9 in non-tumor cells (Table 3C). PD-L1 and LAG-3 staining was not observed in either Ewing sarcoma or osteosarcoma samples. Details of all staining results can be found in Table 4.
| A | Ewing (n=10) | |||||||
| PD-1 | TIM-3 | LAG-3 | ||||||
| + | +/- | - | + | +/- | - | + | +/- | - |
| 1/10 | 3/10 | 6/10 | 10/10 | 0/10 | 0/10 | 0/10 | 0/10 | 10/10 |
| 10% | 30% | 60% | 100% | 0% | 0% | 0% | 0% | 100% |
| PD-L1 | Galectin 9 | MHC Class II | ||||||
| + | +/- | - | + | +/- | - | + | +/- | - |
| 0/10 | 1/10 | 9/10 | 10/10 | 0/10 | 0/10 | 10/10 | 0/10 | 0/10 |
| 0% | 10% | 90% | 100% | 0% | 0% | 100% | 0% | 0% |
| B | Ewing (n=17) | |||||||
| TIM-3 | ||||||||
| + | +/- | - | ||||||
| 17/17 | 0/17 | 0/17 | ||||||
| 100% | 0% | 0% | ||||||
| Galectin 9 | ||||||||
| + | +/- | - | ||||||
| 15/17 | 0/17 | 2/17 | ||||||
| 88.2% | 0% | 11.8% | ||||||
| C | Osteosarcoma n=8 | |||||||
| PD-1 | TIM-3 | LAG-3 | ||||||
| + | +/- | - | + | +/- | - | + | +/- | - |
| 0/8 | 1/8 | 7/8 | 5/8 | 1/8 | 2/8 | 0/8 | 0/8 | 8/8 |
| 0% | 12.5% | 87.5% | 62.5% | 12.5% | 25% | 0% | 0% | 100% |
| PD-L1 | Galectin 9 | MHC Class II | ||||||
| + | +/- | - | + | +/- | - | + | +/- | - |
| 0/8 | 0/8 | 8/8 | 5/8 | 0/8 | 3/8 | 8/8 | 0/8 | 0/8 |
| 0% | 0% | 100% | 62.5% | 0% | 37.5% | 100% | 0% | 0% |
| Ewing (n=10) | ||||||||
| PD-1 | TIM-3 | LAG-3 | ||||||
| + | +/- | - | + | +/- | - | + | +/- | - |
| 1/10 | 3/10 | 6/10 | 10/10 | 0/10 | 0/10 | 0/10 | 0/10 | 10/10 |
| 10% | 30% | 60% | 100% | 0% | 0% | 0% | 0% | 100% |
| PD-L1 | Galectin 9 | MHC Class II | ||||||
| + | +/- | - | + | +/- | - | + | +/- | - |
| 0/10 | 1/10 | 9/10 | 10/10 | 0/10 | 0/10 | 10/10 | 0/10 | 0/10 |
| 0% | 10% | 90% | 100% | 0% | 0% | 100% | 0% | 0% |
| Osteosarcoma n=8 | ||||||||
| PD-1 | TIM-3 | LAG-3 | ||||||
| + | +/- | - | + | +/- | - | + | +/- | - |
| 0/8 | 1/8 | 7/8 | 5/8 | 1/8 | 2/8 | 0/8 | 0/8 | 8/8 |
| 0% | 12.5% | 87.5% | 62.5% | 12.5% | 25% | 0% | 0% | 100% |
| PD-L1 | Galectin 9 | MHC Class II | ||||||
| + | +/- | - | + | +/- | - | + | +/- | - |
| 0/8 | 0/8 | 8/8 | 5/8 | 0/8 | 3/8 | 8/8 | 0/8 | 0/8 |
| 0% | 0% | 100% | 62.5% | 0% | 37.5% | 100% | 0% | 0% |
| NHL n=8 | ||||||||
| PD-1 | TIM-3 | LAG-3 | ||||||
| + | +/- | - | + | +/- | - | + | +/- | - |
| 7/8 | 1/8 | 0/8 | 3/8 | 3/8 | 2/8 | 4/8 | 4/8 | 0/8 |
| 87.5% | 12.5% | 0% | 37.5% | 37.5% | 25% | 50% | 50% | 0% |
| PD-L1 | Galectin 9 | MHC Class II | ||||||
| + | +/- | - | + | +/- | - | + | +/- | - |
| 3/8 | 3/8 | 2/8 | 6/8 | 2/8 | 0/8 | 4/8 | 2/8 | 2/8 |
| 37.5% | 37.5% | 25% | 75% | 25% | 0 | 50% | 25% | 25% |
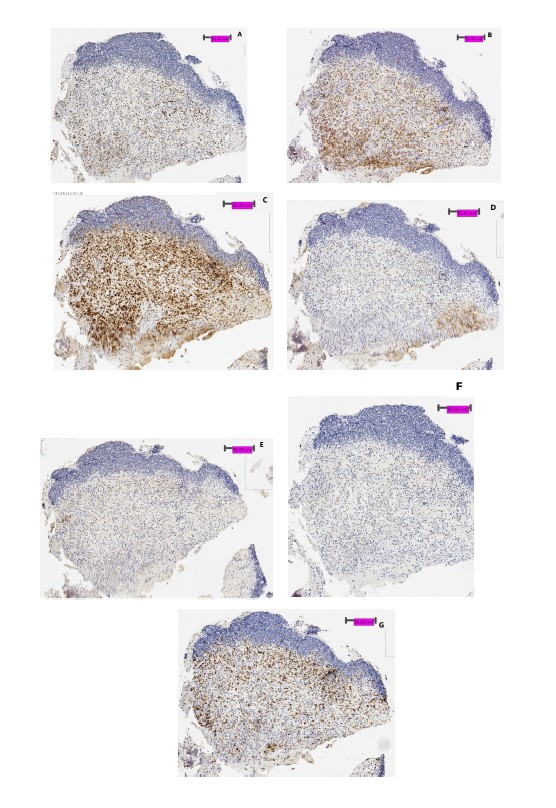
Aside from the differences in overall expression pattern, location of where NCR and ligands were expressed is also noteworthy. We identified a tumor border in 7 of 17 (41%) Ewing sarcoma samples. As represented in Figure 2, staining of checkpoint receptor and ligands was seen in Ewing sarcoma cells congregating along and outside of the tumor border. Few cells were identified within the tumor itself.
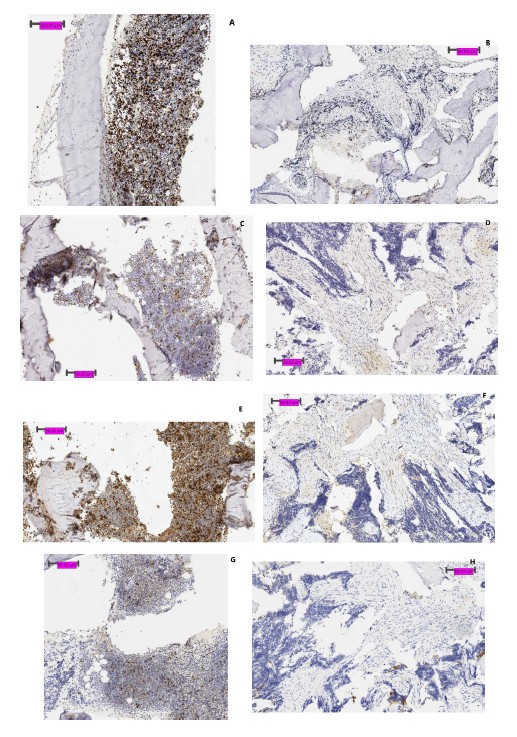
In Figure 3, biopsy specimen includes normal bone marrow in a patient with Ewing sarcoma. Whereas PD-1 stained positive in bone marrow, biopsy of Ewing sarcoma was negative for PD-1 in non-tumor cells. Remainder of staining pattern between tumor and normal tissue appeared to correlate well with each other. Additional staining expression of CD20, 56, and 68 is included in Supplemental data.
No correlation is found between expression of NCR and ligands on tumor and peripheral T cells
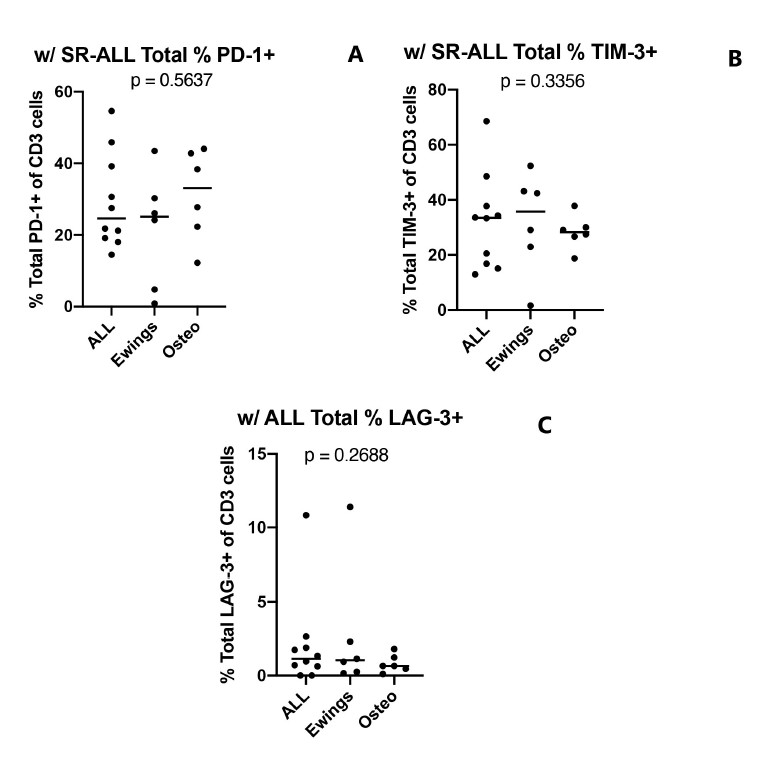
Using standard risk B-cell acute lymphoblastic leukemia (SR B-ALL) as baseline, we examined the expression of receptor and ligands on circulating T cells between the two disease groups and found no statistically significant difference between overall expression of PD-1, TIM-3, or LAG-3 (Figure 4). The reason for choosing SR B-ALL samples as control is due to previously published data that confirmed close resemblance of peripheral T cell subset distribution and biology to healthy children [24-26], as we do not have access to blood from healthy children under these study protocols.
We then compared the expression of NCR and ligands on peripheral T cells to the expression pattern on tumor samples, and no definitive correlation can be made. Whereas TIM-3 and Galectin-9 were highly expressed on non-tumor cells in all Ewing sarcoma biopsies, an increase in TIM-3 expression on peripheral T cells was not found when compared to the osteosarcoma group. Similarly, a lack of PD-1 expression on osteosarcoma tissue samples did not correlate with low expression of PD-1 on peripheral T cells.
T cell expansion potential differs between disease groups at diagnosis and is correlated with total number of NCRs expressed on peripheral T cells
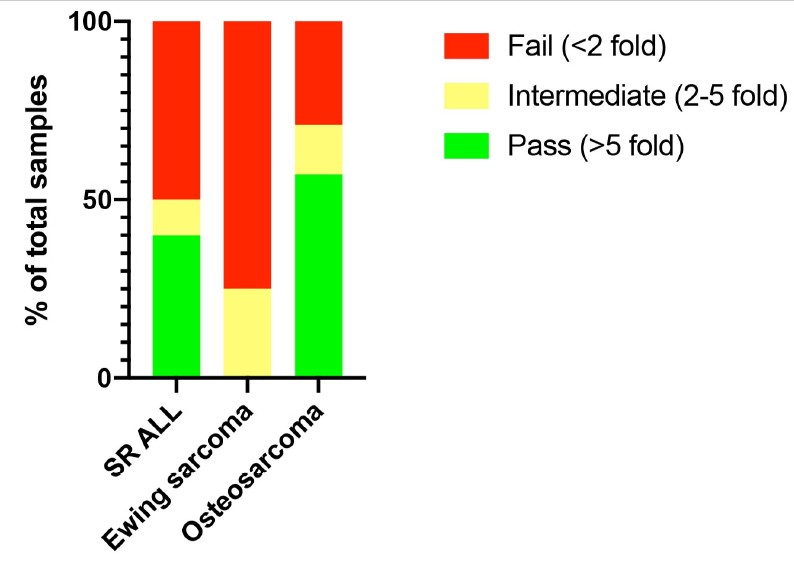
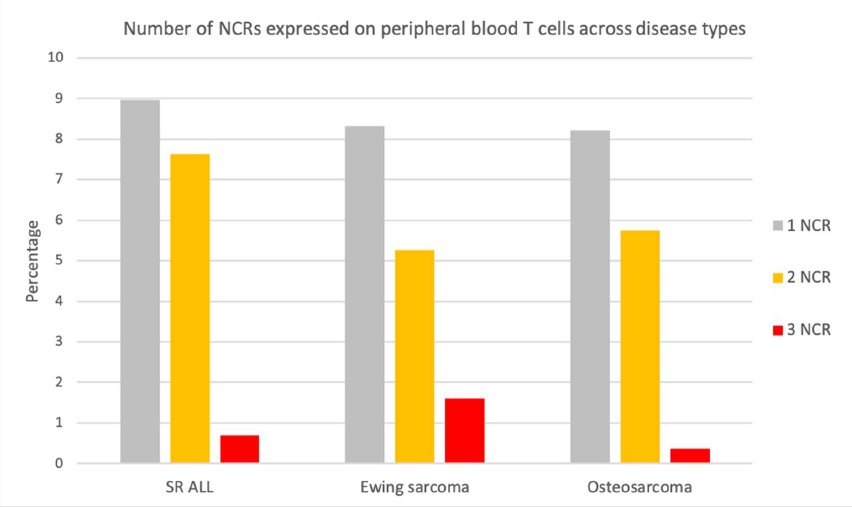
We previously reported a threshold of >5-fold expansion during stimulation with CD3/28 beads was associated with a high likelihood of successful clinical expansion [24]. Therefore, we applied the same threshold to stratify this analysis into samples that “passed” expansion (>5 fold), “failed” expansion (<2 fold), and “indeterminate” (2-5 fold). In this study, SR ALL patients had a higher initial pass rate at diagnosis (40%) than patients with Ewing sarcoma (0%), which is consistent with our previous findings [24]. In contrast, osteosarcoma had a higher initial pass rate than SR-ALL at diagnosis (57%). This is correlated with osteosarcoma patients having the lowest percentage of peripheral T cells that had simultaneous expression of all three NCRs (0.38%), compared to SR ALL (0.7%) and Ewing sarcoma (1.61%) (Table 5, Figure 5).
| 1 NCR | 2 NCR | 3 NCR | |
| SR ALL | 8.97% | 7.63% | 0.7% |
| Ewing sarcoma | 8.31% | 5.27% | 1.61% |
| Osteosarcoma | 8.21% | 5.75% | 0.38% |
Discussion
In this study, we characterized the expression of PD- 1, PD-L1, TIM-3, Galectin 9, LAG-3, MHC Class II on diagnostic tissue samples from patients with Ewing sarcoma and osteosarcoma. Although we observed significant variability in the expression pattern between tumor types, two interesting findings were observed. First, both TIM-3 and Galectin 9 were highly expressed on non-tumor cells in Ewing sarcoma samples at diagnosis. This finding is significant as it can potentially serve as a platform for clinical trials using anti-TIM-3 and anti- Galectin-9 monoclonal antibody in select pediatric solid tumors. However, as few studies to date have evaluated the expression of checkpoint receptors and ligands other than PD-1 and PD-L1, a larger sample size is needed to confirm this finding [27]. Second, osteosarcoma tumor cells at diagnosis did not express any checkpoint receptors and ligands except Galectin-9, and non-tumor cells were also negative for PD-1, PD-L1, and LAG-3, suggesting that these tumors may be resistant to currently available checkpoint inhibitors if used at diagnosis.
The lack of checkpoint receptors and ligands expression on both tumor and non-tumor cells in osteosarcoma diagnostic samples is unique compared to both Ewing sarcoma. In Ewing sarcoma samples, only PD-L1, LAG- 3, and MHC class II were negative on tumor cells, and LAG-3 was also negative on non-tumor cells. Not only does this highlight the variability of the immune-oncologic landscape between different tumor types at diagnosis, it also suggests that up front therapy with checkpoint blockade may not be an ideal choice of therapy for every tumor type.
Despite our findings, it is important to note that the staining pattern observed in our study is different from previously published studies. Factors that contribute to this difference includes the timepoint of which biopsy specimens were obtained: at diagnosis, during treatment, post treatment, or at disease recurrence. In osteosarcoma, expression of checkpoint receptors has been shown to vary extensively [28-31]. Shen et al. found a significant range of PD-L1 expression in osteosarcoma specimens, with 6 of 38 (15.8%) samples having negative expression, 4 of 38 (10.5%) samples with low PD-L1 expression, 19 of 38 (38%) with intermediate PD-L1 expression, and 9 of 38 (23.7%) samples with high PD-L1 expression [30]. However, specimens in this study include diagnostic, treated, and recurrent tumors. Likewise, Chen et al. confirmed that variations in specimen collection time points significantly alter landscape of checkpoint receptors and ligands expression, as PD-1 and PD-L1 were uniformly absent in post-treatment specimens even when its expression were present at diagnosis and in recurrent tumors within the same patient [29]. However, interestingly, even for samples that were all obtained at diagnosis, variations in checkpoint receptors and ligands expression can still be seen. Chen et al. reported PD-1 positivity in 8 of 15 (53%) osteosarcoma samples at diagnosis [29], while Alves et al. noted only “rare” PD-1 positivity in 7 of 21 (33.3%) in diagnostic biopsies of osteosarcoma, with the remainder of the samples having negative expression of PD-1 (14 of 21, 66.6%) [28]. It is important to note that there is no unified immunostaining scoring system between the above studies. Criteria for “positive” and “rare” expression of checkpoint inhibitors varied, with some studies defining “positive” as ≥50% of tumor cells analyzed at 20x magnification, while other studies relied on pathologists’ subjective interpretation of “weak”, “moderate” and “strong” staining patterns [20,29]. The significant variations in expression pattern of checkpoint receptors and ligands across different tumor types points to the importance of a comprehensive investigation of immunohistochemistry staining with uniformity of staining procedures and analysis criteria, while paying close attention to the timepoints from which the specimens were obtained from.
Another important factor contributing to the variation in immunohistochemistry staining pattern lies in the location of where the biopsy was obtained in the tumor. As evidenced by the Ewing sarcoma biopsy sample in Figure 2, non-tumor cells expressing various checkpoint receptors and ligands are congregated at the tumor border, and not dispersed within the tumor. Several other adult studies have described similar findings. Taube et al. demonstrated geographic restriction of PD-L1 and LAG-3 positive tumor cells and non-tumor cells at the tumor border of metastatic melanoma [32]. Aust et al. also reported PD-L1 expression in recurrent serous ovarian tumor was limited to tumor cells at the tumor-stroma border [33]. However, owing to the different biopsy techniques and tumor locations, biopsies that include tumor borders are not often feasible. Of the 25 biopsy samples included in this study, only 7 samples included a tumor border. As a result, expression of checkpoint receptors and ligands may be under reported in samples that did not include a tumor border.
Next, we sought to evaluate the relationship between the expression of checkpoint receptors on peripheral T cells and pediatric tumor biopsy samples at diagnosis. Although expression of TIM-3 and Galectin 9 were seen on most Ewing sarcoma diagnostic biopsy samples, expression of TIM-3 on peripheral T cells at diagnosis was similar to other tumor types. Similarly, although expression of PD-1 and PD-L1 were negative across all osteosarcoma diagnostic biopsy samples, expression of PD-1 on peripheral T cells at diagnosis again did not differ from other tumor types. Our data shows that peripheral blood T cells do not reflect the tumor environment at diagnosis, therefore potentially limiting this parameter’s prognostic value. However, it is interesting to note that a recent study did find a correlation between expression of select checkpoint receptors and TILs. Li et al. looked at a total of 131 adult patients with newly diagnosed breast, kidney, lung, liver, cervical, esophageal, gastric, and colorectal cancer and found that the frequencies of PD-1 and TIM-3 expression in peripheral T cells positively correlated with their respective frequencies in TILs. Other checkpoint receptors such as B and T lymphocyte attenuator (BTLA) and T cell immunoreceptor with Ig and ITIM domains (TIGIT) were not shown to have statistically significant correlation between peripheral T cells and TILs [34]. Although the difference in our results may be a reflection of the limited number of patients in our study, it is also possible that different tumors impart a different effect on peripheral T cells as the tumors evaluated in Li et al. did not include Ewing sarcoma, osteosarcoma, or NHL samples. Furthermore, pediatric tumors may have unknown intrinsic biologic differences compared to adult tumors (i.e. carcinomas) which contribute to varying oncoimmunologic landscapes, highlighting the importance of future similar studies in order to build upon our current understanding of this intricate relationship.
Although the expression profile of checkpoint receptors appear to be similar across the tumor types, qualitative differences were observed through differences in vivo expansion potential. Using a threshold of >5-fold T cell expansion during stimulation with CD3/CD28 beads as a marker for “healthy” effector function, we demonstrate that 57% of osteosarcoma samples in this study had >5- fold T cell expansion, compared to 0% of Ewing sarcoma (Figure 6). This finding reassuringly confirms our prior work that demonstrates a higher pass rate in diagnostic osteosarcoma than Ewing sarcoma samples [24]. This may in part be due to the distribution of peripheral T cell subsets with osteosarcoma patients having more naïve and stem central memory T cells than Ewing sarcoma patients [24], as well as a relative preservation of T cell effector function as only 0.38% of total peripheral T cells in osteosarcoma samples had co-expression of all three checkpoint receptors at diagnosis (Table 5, Figure 5). This finding again supports that different tumor microenvironments may be associated with global immune changes by varying degrees and mechanisms.
Limitations of this study includes small sample size within each tumor group which may contribute to the variations in IHC staining results seen in our study compared with prior studies. IHC staining results may also be affected by the decalcification process of bone tumors, as well as crush artifacts in biopsy specimens. We are at risk of observational bias by working with only one pathologist, and interpretation subjectivity remains to be a barrier that is difficult to overcome. Use of different antibodies against checkpoint receptors and ligands, along with different staining techniques further complicate results. Sampedro-Nunez et al. reported the use of three different antibodies against PD-L1 but only one antibody stained positive for PD-L1 in their tumor samples [35]. Finally, the lack of biopsy samples that included a tumor border may also underestimate the true incidence of checkpoint receptors and ligands expression found within and around the tumor.
In summary, we provide the first ever study that describe the comprehensive onco-immunologic landscape in pediatric solid tumors by correlating expression of checkpoint receptor and ligand expression on treatment naïve biopsy samples with peripheral blood T cells. We demonstrate high expression of TIM-3 and Galectin 9 positivity on immunohistochemistry staining in Ewing sarcoma, along with a relative lack of checkpoint receptor and ligand expression in osteosarcoma samples. However, such findings on biopsy samples are not reflected in peripheral blood T cells therefore suggesting a more complex interaction between the tumor microenvironment and the global immune system as well as highlighting the limitations of peripheral blood biomarkers in this setting. Further understanding is urgently needed in order to maximize the clinical benefit of checkpoint inhibitors in pediatric solid tumors.
Funding
This work is supported by The Doris Duke Charitable Foundation (DMB) and Jeffrey Pride Foundation (DMB).
References
2. Decker WK, da Silva RF, Sanabria MH, Angelo LS, Guimarães F, Burt BM, et al. Cancer immunotherapy: historical perspective of a clinical revolution and emerging preclinical animal models. Frontiers in Immunology. 2017 Aug 2;8:829.
3. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New England Journal of Medicine. 2014 Oct 16; 371(16):1507-17.
4. Davis KL, Fox E, Merchant MS, Reid JM, Kudgus RA, Liu X, et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): A multicentre, open-label, single-arm, phase 1–2 trial. The Lancet Oncology. 2020 Apr 1; 21(4):541-50.
5. Wagner LM, Adams VR. Targeting the PD-1 pathway in pediatric solid tumors and brain tumors. OncoTargets and therapy. 2017; 10:2097.
6. Wherry EJ. T cell exhaustion. Nature Immunology. 2011 Jun; 12(6):492-9.
7. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nature Reviews Immunology. 2015 Aug; 15(8):486-99.
8. Gorris MA, Halilovic A, Rabold K, van Duffelen A, Wickramasinghe IN, Verweij D, et al. Eight-color multiplex immunohistochemistry for simultaneous detection of multiple immune checkpoint molecules within the tumor microenvironment. The Journal of Immunology. 2018 Jan 1; 200(1):347-54.
9. Madore J, Vilain RE, Menzies AM, Kakavand H, Wilmott JS, Hyman J, et al. PD-L1 expression in melanoma shows marked heterogeneity within and between patients: implications for anti-PD-1/PD-L 1 clinical trials. Pigment Cell & Melanoma Research. 2015 May; 28(3):245-53.
10. Mansfield AS, Murphy SJ, Peikert T, Eunhee SY, Vasmatzis G, Wigle DA, et al. Heterogeneity of programmed cell death ligand 1 expression in multifocal lung cancer. Clinical Cancer Research. 2016 May 1; 22(9):2177-82.
11. Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Science Translational Medicine. 2012 Mar 28; 4(127):127ra37-.
12. Lyford-Pike S, Peng S, Young GD, Taube JM, Westra WH, Akpeng B, et al. Evidence for a role of the PD-1: PDL1 pathway in immune resistance of HPV-associated head and neck squamous cell carcinoma. Cancer Research. 2013 Mar 15; 73(6):1733-41.
13. Darvin P, Toor SM, Nair VS, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Experimental & Molecular Medicine. 2018 Dec; 50(12):1-1.
14. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. New England Journal of Medicine. 2010 Aug 19; 363(8):711-23.
15. Merchant MS, Wright M, Baird K, Wexler LH, Rodriguez-Galindo C, Bernstein D, et al. Phase I clinical trial of ipilimumab in pediatric patients with advanced solid tumors. Clinical Cancer Research. 2016 Mar 15; 22(6):1364-70.
16. Shindiapina P, Alinari L. Pembrolizumab and its role in relapsed/refractory classical Hodgkin’s lymphoma: evidence to date and clinical utility. Therapeutic Advances in Hematology. 2018 Apr;9(4):89-105.
17. Paydas S, Bagir EK, Deveci MA, Gonlusen G. Clinical and prognostic significance of PD-1 and PD-L1 expression in sarcomas. Medical Oncology. 2016 Aug; 33(8):1-0.
18. Kim C, Kim EK, Jung H, Chon HJ, Han JW, Shin KH, et al. Prognostic implications of PD-L1 expression in patients with soft tissue sarcoma. BMC Cancer. 2016 Dec; 16(1):1-7.
19. Inaguma S, Wang Z, Lasota J, Sarlomo-Rikala M, McCue PA, Ikeda H, et al. Comprehensive immunohistochemical study of programmed cell death ligand 1 (PD-L1). Analysis in 5536 cases revealed consistent expression in trophoblastic tumors. The American Journal of Surgical Pathology. 2016 Aug; 40(8):1133.
20. van Erp AE, Versleijen-Jonkers YM, Hillebrandt Roeffen MH, van Houdt L, Gorris MA, van Dam LS, et al. Expression and clinical association of programmed cell death-1, programmed death-ligand-1 and CD8+ lymphocytes in primary sarcomas is subtype dependent.Oncotarget. 2017 Sep 19; 8(41):71371.
21. Majzner RG, Simon JS, Grosso JF, Martinez D, Pawel BR, Santi M, et al. Assessment of programmed deathligand 1 expression and tumor-associated immune cells in pediatric cancer tissues. Cancer. 2017 Oct 1; 123(19):3807- 15.
22. Geoerger B, Karski EE, Zwaan M, Casanova M, Marshall LV, DuBois SG, et al. A phase I/II study of atezolizumab in pediatric and young adult patients with refractory/ relapsed solid tumors (iMATRIX-Atezolizumab). Journal of Clinical Oncology; 2017.
23. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nature Immunology. 2009 Jan; 10(1):29-37.
24. Das RK, Vernau L, Grupp SA, Barrett DM. Naive T-cell deficits at diagnosis and after chemotherapy impair cell therapy potential in pediatric cancers. Cancer Discovery. 2019 Apr 1;9(4):492-9.
25. Singh N, Perazzelli J, Grupp SA, Barrett DM. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Science Translational Medicine. 2016 Jan 6; 8(320):320ra3-.
26. Shearer WT, Rosenblatt HM, Gelman RS, Oyomopito R, Plaeger S, Stiehm ER, et al. Lymphocyte subsets in healthy children from birth through 18 years of age: the Pediatric AIDS Clinical Trials Group P1009 study. Journal of Allergy and Clinical Immunology. 2003 Nov 1; 112(5):973-80.
27. Mochizuki K, Kawana S, Yamada S, Muramatsu M, Sano H, Kobayashi S, et al. Various checkpoint molecules, and tumor-infiltrating lymphocytes in common pediatric solid tumors: Possibilities for novel immunotherapy. Pediatric Hematology and Oncology. 2019 Jan 2; 36(1):17- 27.
28. Alves PM, de Arruda JA, Arantes DA, Costa SF, Souza LL, Pontes HA, et al. Evaluation of tumor-infiltrating lymphocytes in osteosarcomas of the jaws: a multicenter study. Virchows Archiv. 2019 Feb; 474(2):201-7.
29. Chen S, Guenther LM, Aronhalt A, Cardillo L, Janeway KA, Church AJ. PD-1 and PD-L1 expression in osteosarcoma: Which specimen to evaluate?. Journal of pediatric Hematology/Oncology. 2020 Nov 11; 42(8):482- 7.
30. Shen JK, Cote GM, Choy E, Yang P, Harmon D, Schwab J, et al. Programmed cell death ligand 1 expression in osteosarcoma. Cancer Immunology Research. 2014 Jul 1; 2(7):690-8.
31. Chowdhury F, Dunn S, Mitchell S, Mellows T, Ashton- Key M, Gray JC. PD-L1 and CD8+ PD1+ lymphocytes exist as targets in the pediatric tumor microenvironment for immunomodulatory therapy. OncoImmunology. 2015 Oct 3; 4(10):e1029701.
32. Taube JM, Young GD, McMiller TL, Chen S, Salas JT, Pritchard TS, et al. Differential expression of immune-regulatory genes associated with PD-L1 display in melanoma: implications for PD-1 pathway blockade. Clinical Cancer Research. 2015 Sep 1; 21(17):3969-76.
33. Aust S, Felix S, Auer K, Bachmayr-Heyda A, Kenner L, Dekan S, et al. Absence of PD-L1 on tumor cells is associated with reduced MHC I expression and PD-L1 expression increases in recurrent serous ovarian cancer. Scientific Reports. 2017 Mar 7; 7(1):1-2.
34. Li X, Wang R, Fan P, Yao X, Qin L, Peng Y, et al. A comprehensive analysis of key immune checkpoint receptors on tumor-infiltrating T cells from multiple types of cancer. Frontiers in Oncology. 2019 Oct 25; 9:1066.
35. Sampedro-Núñez M, Serrano-Somavilla A, Adrados M, Cameselle-Teijeiro JM, Blanco-Carrera C, Cabezas- Agricola JM, et al. Analysis of expression of the PD-1/PDL1 immune checkpoint system and its prognostic impact in gastroenteropancreatic neuroendocrine tumors. Scientific Reports. 2018 Dec 13;8(1):1-1.