Abstract
Peripheral circulating B-lymphocytes and B-lymphocytes in the bone marrow (BM) show different responses to lymphotoxic or immunosuppressive agents. We explored the existence of a dysregulated distribution of B-lymphocytes between peripheral and BM compartments and the underlying mechanisms. The percentage of CXC chemokine receptor 4+ B (CXCR4+ B) cells was decreased in the peripheral blood (PB) and increased in the BM of MRL/lpr mice and SLE patients. CXC chemokine ligand 12 (CXCL12) production by BM osteoblasts (OBs) derived from MRL/lpr mice and SLE patients was higher than that obtained with C57BL/6 mice or healthy subjects. MRL/lpr-derived OBs demonstrated stronger chemotactic ability toward B-lymphocytes than control OBs, and more B-lymphocytes colocalized with OBs within the periosteal zone in MRL/lpr mice. Moreover, the CXCR4+ B cell percentages were negatively correlated with the serum immunoglobulin G concentration, and the BM CXCL12 levels were positively correlated with the systemic lupus erythematosus disease activity index score and anti-double stranded DNA titer and negatively correlated with the serum complement 3 concentration. In conclusion, our results indicate a shifted distribution of B-lymphocytes between the BM and peripheral compartments in SLE patients and MRL/lpr mice and that the upregulation of CXCL12 in OBs likely contributes to enhanced chemotactic migration and anchorage of B-lymphocytes to OBs.
Keywords
SLE, CXCL12, B cells, Bone marrow, Osteoblasts
Introduction
Systemic lupus erythematosus (SLE) is a multiorgan, multisystem autoimmune disease characterized by the production of autoantibodies, a lack of immunological tolerance, increased levels of inflammatory cytokines, and antibody removal disorders, among other features, which lead to a series of chain reactions that damage target organs and thus cause diseases [1]. B-lymphocytes occupy a central position in the promotion of autoimmunity in SLE and mediate disease not only by producing autoantibodies (Abs) but also by expanding autoreactive CD4+ T cells via autoantigen (Ag) presentation through a positive feedback loop with CD4+ T-lymphocytes [2]. The abnormal activation of multiclonal B-lymphocytes may result from inherent high reactivity, ineffective negative selection, deficits in immunomodulation, and an overactive inflammatory environment, among other factors [3]. Considering the important role of B-cell abnormalities in SLE, anti-CD20 monoclonal antibodies such as rituximab have been used with success in the management of refractory SLE. However, the benefits of the addition of rituximab to the treatment of SLE patients remain controversial, and some clinical trials have failed to demonstrate the superiority of rituximab to conventional immunosuppressive therapy. In addition, SLE patients initially responding to rituximab treatment may develop resistance to subsequent rituximab administration [4,5]. All these factors suggest the complexity of abnormal B-cell-mediated immunity in SLE.
A previous study found that an abnormal CXC chemokine receptor 4/CXC chemokine ligand 12 (CXCR4/CXCL12) axis plays an important role in the pathogenesis of lupus. For example, CXCR4 is overexpressed in immune cells such as B cells, monocytes and neutrophils, which enhances the migration of immune cells toward the CXCL12 gradient and thereby facilitates cell survival and plays a pivotal role in disease development [6]. CXCL12, as the ligand for CXCR4, can activate a variety of cell signaling pathways by interacting with its receptor, and these pathways include mechanistic target of rapamycin (mTOR), phosphatidylinositol 3 kinase/protein kinase B (PI3K/AKT), nuclear factor kappa-B (NF-κB) and Janus kinase/signal transduction and activator of transcription (JAK/STAT) pathways [7,8]. Some studies have shown that the levels of CXCL12 in the serum and kidneys of SLE patients are higher than those in controls and correlated with disease activity [9,10]. Although the levels of CXCR4 on antibody-secreting cells are similar between SLE patients and healthy controls [11], some lupus models with active nephritis, such as NZBWF1/J and MRL/lpr mice, have shown conflicting results indicating that the expression of CXCR4 on several PB leukocyte subsets is upregulated [6,12,13], which could prolong the B-cell lifespan and augment B-cell chemotaxis. Studies have shown that knockout of CXCL12 is lethal during fetal development in mice and induce a significant reduction in B-lymphocytes in the bone marrow (BM) [14,15] because CXCL12 is involved in the migration of B cells to the BM [16]. In addition to B-cell development, the CXCR4/CXCL12 axis is also reportedly involved in biological processes such as angiogenesis, inflammation, and cancer metastasis [17,18].
Our previous studies identified multiple abnormalities within the BM microenvironment in SLE subjects, especially deficit of BM-derived mesenchymal stem cells (MSCs) [19,20], which are the progenitor for osteoblasts (OBs). In addition to bone formation, OBs participate in the regulation of hematopoiesis within the BM via the CXCR4/CXCL12 axis [21]. In SLE, whether the CXCR4/CXCL12 axis between OBs and B-lymphocytes is involved in disease pathogenesis is currently unclear. To address this knowledge gap, we investigated the CXCR4/CXCL12 axis between OBs and B-lymphocytes, especially its role in the regulation of the distribution of B-lymphocytes within the BM and peripheral compartments.
Materials and Methods
Human samples
Patients diagnosed with SLE according to the 2012 American College of Rheumatology classification criteria and healthy controls were recruited from the Affiliated Hospital of Jiangsu University. All experiments with human samples were approved by the Ethics Committee of the Affiliated Hospital of Jiangsu University (reference number: KY2021K0909). The demographic information of the SLE patients and healthy controls is shown in Supplementary Tables S1 and S2.
Mice
Female MRL/lpr mice (specific pathogen-free (SPF) grade) and female C57/BL6 mice (SPF grade) (both purchased from Shanghai Slack Experimental Animals Limited Liability Company, license number: SCXK (Shanghai) 2017-0005) were fed in the SPF-grade breeding area of the Animal Experimental Center of Jiangsu University. All animals were fed until 12-14 weeks, when the experiment was initiated. All animal experiments were approved by the Animal Experiment Committee of Jiangsu University and conducted in accordance with institutional and national guidelines. All animal experiments were approved by the Animal Ethics Committee of the Jiangsu University Animal Laboratory Center (reference number: UJS-IACUC-AP-2020032539).
Flow cytometry
BM cells were harvested from the femurs and tibias of mice, and PB mononuclear cells were harvested from the inner orbital vein of the mice and processed to obtain single-cell suspensions. Then, 1 x10^7/mL cells were incubated in 100 µL of dye buffer (phosphate buffered saline (PBS) and 2% fetal bovine serum (FBS)) with antibodies. PB cells were stained with anti-mouse CD19-PE (clone 1D3; BD Biosciences) and anti-CXCR4-FITC (ab1670, ab150129), and BM cells were stained with anti-B220-PE (cloneRA3-6B2; BD Biosciences) and anti-CXCR4-FITC (ab1670, ab150129). We collected clinical waste BM samples from SLE patients and healthy individuals without autoimmune diseases who underwent lumbar vertebral surgery to obtain BM cells and their supernatants. BM cells were stained with anti-human CD19-PE (clone HIB19; BD Biosciences) and anti-CXCR4-FITC (ab1670, ab150129). All flow cytometric data were acquired using a BD LSRFortessa cytometer (BD Biosciences) and analyzed with FlowJo software (BD Biosciences).
RNA extraction and RT?qPCR
The BM, spleen, and PB of mice were obtained and processed into single-cell suspensions. RNA was extracted with an RT reagent kit reverse transcription kit (TaKaRa) according to the manufacturer’s instructions. Gene expression was quantified with a Takara STBR Premix Ex Taq kit and a Q5 qRT?PCR instrument (ABI, USA). The primers were as follows: CXCL12 sense, 5'-TCTGAAAATCCTCAACACTCCA-3', and anti-sense, 5'-CAGGTACTCTTGGATCCACTTT-3'; and β-actin sense, 5'-GTGCTATGTTGCTCTAGACTTCG-3', and anti-sense, 5'-ATGCCACAGGATTCCATACC-3'.
ELISA
The CXCL12 levels in serum, BM supernatant and culture medium supernatant samples were measured using a human CXCL12 ELISA kit (Multi Sciences) and mouse CXCL12 ELISA kit (Multi Sciences). All samples were treated and diluted equally before ELISA. Sandwich ELISAs for human and mouse CXCL12 were performed according to the manufacturer’s instructions (Multi Sciences).
Cell culture
OB culture: Murine OBs were obtained from published procedures with minor modifications [22]. In brief, the calvariae were removed aseptically from 12-week-old C57 or MRL/lpr mice and subjected to repeated digestion for 10 minutes at 37°C with 0.25% type II collagenase (Millipore) in PBS, and the supernatant was then discarded. The bone fragments were added to 0.1% type II collagenase for repeated digestion for 10 minutes at 37°C, and the supernatant was discarded. Subsequently, the bone fragments were digested with 0.1% type II collagenase for 20 minutes, and the supernatant was collected; these steps were repeated four times. Fractions were then collected by centrifugation and cultured in MEM Alpha Modification (α-MEM) containing 10% FBS and 1% penicillin?streptomycin. The cells were cultured to 80% confluence (undifferentiated OBs). In some experiments, the cells were then cultured in differentiation medium (α-MEM containing 10% FBS, 0.05 mmol/L ascorbic acid, 100 mmol/L dexamethasone and 10 mmol/L β-glycerophosphate) for 1 or 2 weeks (differentiated OBs) to obtain OBs-1w and OBs-2w, respectively. The differentiated OBs (OBs2W) were then used for in vitro migration assays.
BM mesenchymal stem cell culture
Mice (aged 12-14 weeks) were anesthetized, and their tibias and fibulas were dissected after disinfection. The BM was repeatedly flushed with Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Waltham, MA, USA) and then centrifuged at 1000 rpm for 10 min. The resulting pellet was collected and resuspended in DMEM containing 10% FBS (Gibco) and 1% penicillin?streptomycin (Gibco) in a 100-mm culture dish, which was then placed in a 37°C humidified incubator with a 5% CO2 atmosphere. Approximately 3 days later, the cells were confluent in the culture dish, and these cells were designated as the first cell culture passage. The cells were then subpassaged at a ratio of 1:1, expanded, purified, and cultured to generate third-passage cells.
In vitro migration assays
B cells were obtained from the spleens of C57 or MRL/lpr mice by magnetic cell sorting (MACS-STEMCELL). The migration of splenic B-cell populations was evaluated in 24-well CoStar Transwell chambers (5-µm pore size, Corning). Briefly, a suspension of 107 B cells in 100 µL of Roswell Park Memorial Institute 1640 (RPMI 1640) medium without FBS was added to each upper chamber, and each lower chamber was seeded with OBs or OBs infected with lentiviral short hairpin RNA (shRNA) 340 at the same density. These OBs were osteogenically induced two weeks before the experiment. In some experiments, a CXCL12 neutral ligand (LIT-927, SELLECK) dissolved in dimethyl sulfoxide (DMSO) (66 mg/mL) was added to the lower chamber prior to migration. After 24 h at 37°C, the B cells in suspension in the lower chamber were harvested and counted with a blood cell counter. The results are expressed as the numbers of B cells that migrated to the lower chamber.
Lentiviral infection
Lentiviruses were packaged and produced according to the manufacturer’s instructions (Gene Pharma). The gene names of the lentiviruses were LV3-CXCL12-Mus-340 (5-CCCGAAATTAAAGTGGATCCA-3) and LV3-NC (5-TTCTCCGAACGTGTCACGT-3). OBs (1x105) were split into a 24-well plate and grown to 60-80% confluence after 24 h. The plates were refed medium for OBs, specifically 400 µL of α-MEM (Gibco) without FBS, 2 h prior to transfection. Then, 100 µL of lentivirus solution was added to each well. Twenty-four hours after transfection, the medium was replaced with fresh complete medium (10% α-MEM). The fluorescence (GFP) of OBs was observed under a fluorescence microscope at 72 h after transfection, and GFP-positive OBs were isolated by fluorescence-activated cell sorting (FACS). Subsequently, GFP-positive OBs were induced to undergo osteogenic differentiation for 2 weeks and used for in vitro migration assays. The inhibitory efficiency of shRNA340 against CXCL12 mRNA was monitored by RT?PCR.
B-cell homing assay
CD19+ B cells were recovered from the spleens of MRL/lpr mice by positive selection using a MACS microbead isolation kit (STEMCELL) according to the manufacturer’s instructions. The cells were then resuspended at 1x108 cells/mL in PBS, an equal volume of carboxyfluorescein diacetate succinimidyl ester (CFDA-SE, STEMCELL) was added as a 2X working stock solution to obtain a final concentration of 0.5-10 µM, and the cells were incubated with the dye for 5-10 minutes in the dark at room temperature. After washing, approximately 1x108 CFDA-stained CD19+ B cells were injected retro-orbitally into 12- to 13-week-old syngeneic C57BL/6 mice, MRL/lpr mice and MRL/lpr mice pretreated with LIT-927 (ig, 10 mg/kg) for 12 h. The percentage of CFDA-labeled cells in the cell mixture was determined by flow cytometry before injection to measure the number of CFDA-labeled cells transferred. The recipient mice were killed 24 h after injection, and the percentage of CFDA-labeled B cells in the BM was analyzed by flow cytometry.
Immunofluorescence assay
Bones from sacrificed C57BL/6 and MRL/lpr mice were cut into paraffin sections and rehydrated. After antigen retrieval and endogenous peroxidase blockade, we covered the objective tissues with 10% donkey serum or 3% bovine serum albumin (BSA) at room temperature for 30 min. The antibodies used for the immunolabeling of BM sections were anti-B220 (cloneRA3-6B2; BD Biosciences), anti-CXCL12 (GB11624; Servicebio), anti-CD31 (GB12063; Servicebio), and anti-CD51 (GB11293-2; Servicebio). The cells were then incubated with 4’,6-diamidino-2’-phenylindole (DAPI) solution at room temperature for 10 min, kept in the dark and cover slipped with antifade mounting medium. Images were collected by fluorescence microscopy in conjunction with a Nikon imaging system. DAPI emits blue fluorescence (emission wavelength: 420 nm) in response to an ultraviolet (UV) excitation wavelength of 330-380 nm, fluorescein isothiocyanate (FITC) emits green fluorescence (emission wavelength: 515-555 nm) in response to an excitation wavelength of 465-495 nm, and cyanine 3 (CY3) emits red fluorescence (emission wavelength: 590 nm) in response to an excitation wavelength of 510-560 nm. Differences between the expression of CXCL12 between C57 and MRL/lpr BM have been analysed by Image J.
Statistical analysis
All analyses were performed using GraphPad Prism version 8 (USA). The data are expressed as the means ± SDs and were compared by independent Student’s t test or a nonparametric test. The correlation between the percentages of CXCR4+ B cells or the level of CXCL12 in BM and markers of disease activity in patients with SLE was measured by Pearson correlation coefficients. P value <0.05 was considered to indicate statistical significance.
Results
The BM of MRL/lpr mice and SLE patients exhibited an increased percentage of CXCR4+ B cells and higher levels of CXCL12
First, the percentages of CXCR4+ B cells in the PB or BM were determined by flow cytometry. Consistent with the findings reported by Wang et al. (Wang et al., 2009), a lower percentage of CXCR4+ B-lymphocytes in the PB and a higher percentage of these B-lymphocytes in the BM were found in MRL/lpr mice compared with C57 mice (Figures 1A and 1B). Because CXCL12 is the ligand for CXCR4, we subsequently examined the CXCL12 levels in the BM, spleen and PB of MRL/lpr mice. The levels of CXCL12 in both the BM and PB were markedly higher in MRL/lpr mice than in C57 control mice, and the highest CXCL12 level was detected in the BM among the three sites (Figure 1C). Similar to the results found in lupus mice, a higher percentage of CXCR4+ B cells and higher levels of CXCL12 were detected in the BM of SLE patients than in that of healthy controls, although the difference in the CXCL12 levels was not significant (Figures 1D and 1E).
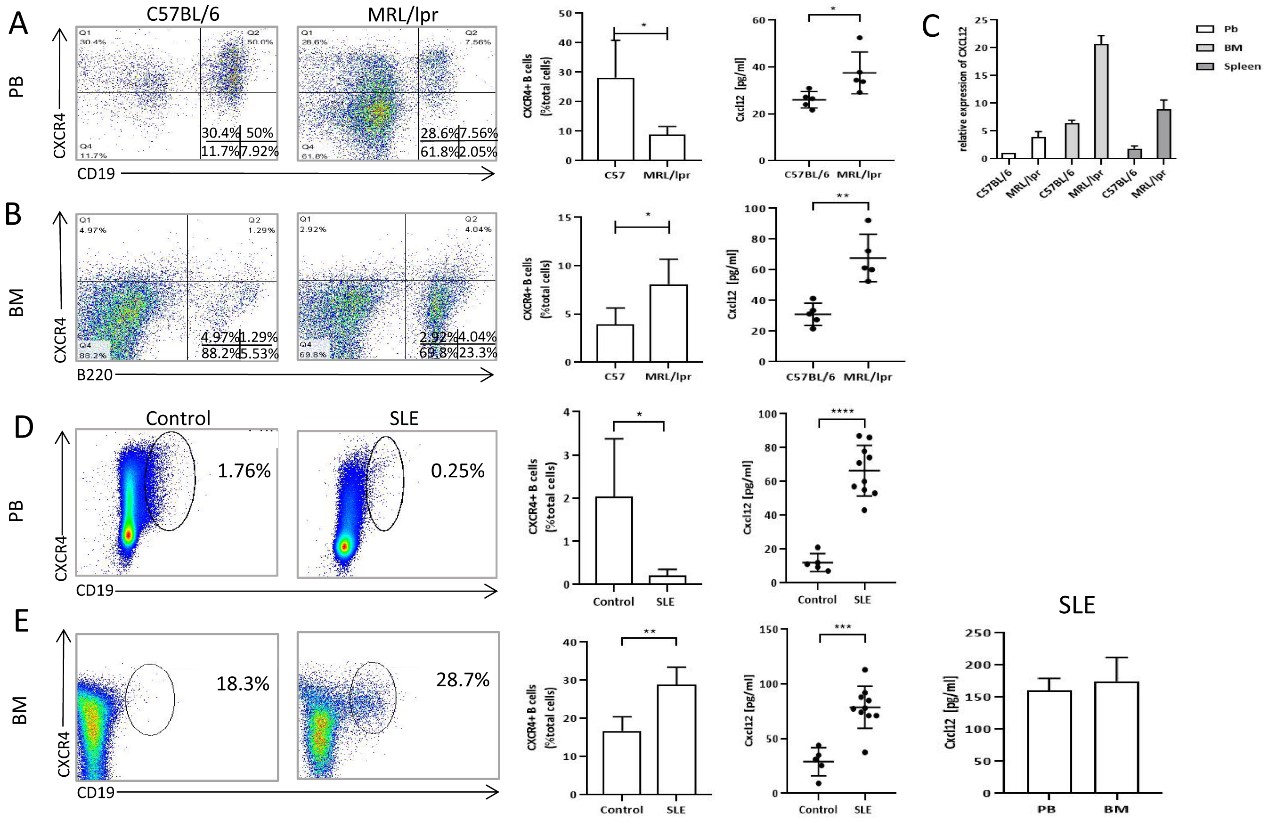
Figure 1. Differences in the percentage of CXCR4+ B cells and the levels of CXCL12 between control and SLE subjects. A: The percentages of CXCR4+B cells in peripheral blood of MRL/lpr mice (n=5) and C57 mice (n=5) measured by flowmetry, and levels of CXCL12 in peripheral blood of MRL/lpr mice (n=5) and C57 mice (n=5) by ELISA. B: The percentages of CXCR4+ B cells in bone marrow of MRL/lpr mice (n=5) and C57 mice (n=5) measured by flowmetry, and levels of CXCL12 in bone marrow of MRL/lpr mice (n=5) and C57 mice (n=5) measured by ELISA. C: mRNA expression of CXCL12 in peripheral blood, bone marrow, spleen of MRL/lpr mice (n=5) and C57 mice (n=5) measured by qRT-PCR. D: The percentages of CXCR4+ B cells and expression of CXCL12 protein in peripheral blood of healthy controls (n=5) and SLE (n=10). E: The percentages of CXCR4+ B cells and expression of CXCL12 protein in bone marrow of healthy controls (n=5) and SLE (n=10). The last panel is the expression of CXCL12 between PB and BM of SLE (n=5). Data represented the mean ± SD. *P<0.05, ** P<0.01, *** P<0.001, ****P<0.0001, compared with C57BL/6 (A, B, C) and control (D, E).
MRL/lpr-derived OBs produced more CXCL12 than BMMSCs
Reportedly, many types of cells in the BM stroma, including MSCs, endothelial cells and OBs, produce CXCL12 [23]. We then successfully obtained OBs from the mouse skull, and their osteogenic potential was validated by alkaline phosphatase (ALP) staining (Figure 2A). Compared to MSCs, OBs were shown to produce higher levels of CXCL12 (Figure 2B). In addition, MRL/lpr-derived OBs were also shown to produce higher levels of CXCL12 than C57-derived cells after 2w culture with osteogenic medium (Figure 2C).
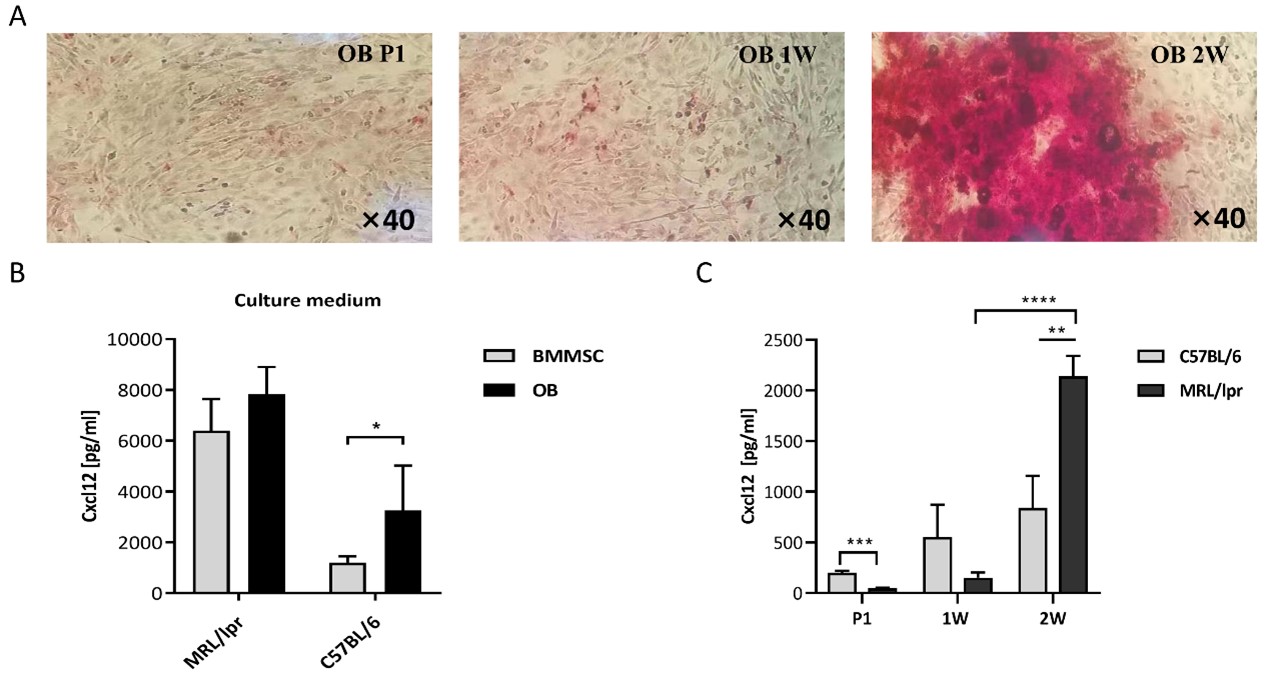
Figure 2. Expression of CXCL12 protein from mice OBs. A: Alkaline phosphatase staining before and after the implementation of osteogenic medium. B: Expression of CXCL12 in BMMSCs and OBs from C57 and MRL/lpr mice (n=5). C: The change of CXCL12 levels in the supernatant of the culture during osteogenic induction. CXCL12 levels were detected by ELISA at the beginning, 1 week (1W) and 2 weeks (2W) of induction. Data represented the mean ± SD. *P<0.05, **P<0.01, ***P< 0.001, ****P<0.0001, compared with C57.
CXCL12 mediated enhanced chemotactic migration of B-lymphocytes to OBs in SLE
To determine the possible mechanism responsible for the higher percentage of CXCR4+ B cells in the BM of SLE patients, we first investigated the capacity of hematopoietic stem cells (HSCs) to differentiate into B-lymphocytes. We found that the number of CFU-pre-B from MRL/lpr mice was lower than that from C57 mice, which does not support a finding of enhanced B-lymphomagenesis in the BM of SLE mice [S3]. Based on this result, we hypothesized that CXCR4+ B-cell migration to and retention in the BM were likely enhanced and that aberrant chemotaxis plays an important role in this process.
We subsequently investigated the chemotactic migration of B-lymphocytes by a transwell assay. B cells from C57 mice showed greater migration to MRL/lpr-derived OBs than to C57-derived OBs (Figure 3B). This observation implied that MRL/lpr-derived OBs produced more potent and/or higher levels of chemokines to attract B-lymphocytes. To confirm the role of CXCL12 in the chemotactic effect of OBs, we then transfected C57-derived or MRL/lpr-derived OBs with shRNA constructed by lentiviral vectors to downregulate CXCL12 expression. As shown in Figure 3A, CXCL12-shRNA transfection could effectively reduce the level of CXCL12 in the supernatant of OB cultures, as revealed by ELISA. In a subsequent study, we employed CXCL12-shRNA-transfected OBs as coating cells in the lower chamber. Compared with nontransfected cells, the migration of B-lymphocytes toward OBs was significantly suppressed, regardless of whether the OBs were derived from C57 or MRL/lpr mice. Furthermore, the addition of LIT-927 (a neutraligand of CXCL12) to the transwell decreased the migration of both C57- and MRL/lpr-derived B-lymphocytes. In addition, its inhibition of chemotactic migration was shown to occur in a dose-dependent manner (Figures 3C and 3D).
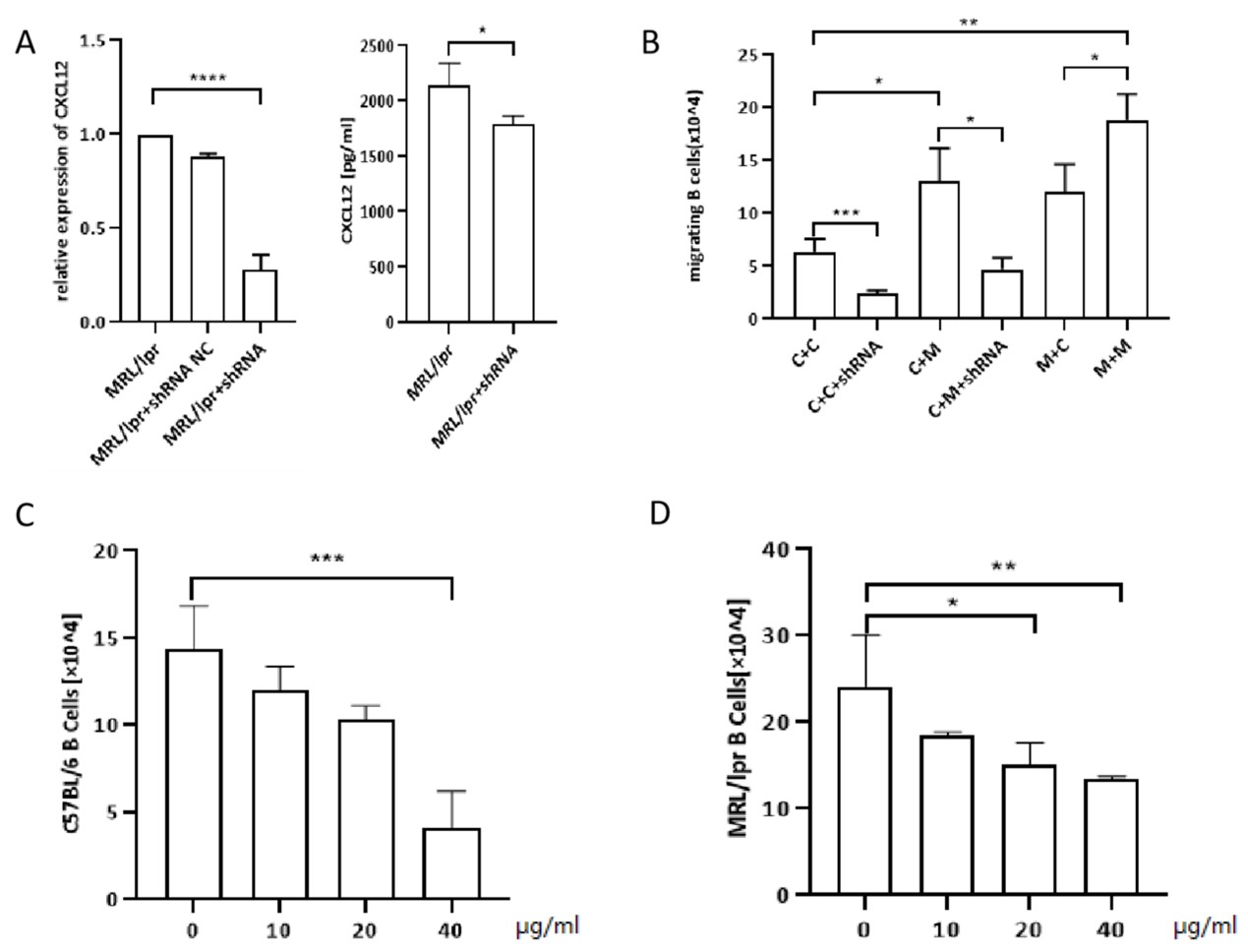
Figure 3. The number of transwell migration of B cells to OBs. A: shRNA efficiency was shown by qRT-PCR. shRNA effectively decreases supernatant concentration of CXCL12 in the OBs culture.(MPL/lpr+shRNA: MPL/lpr-derived OBs transinfected with shRNA by lentiviral vector ) B: Transwell migration of splenic B cells (upper chamber) to OBs (lower chamber): C+C: C57/BL6 mice B cells (upper chamber) + C57/BL6 mice OBs (lower chamber); C+C+shRNA: C57/BL6 mice B cells (upper chamber) + C57/BL6 mice OBs transfected with shRNA (lower chamber); C+M: C57/BL6 mice B cells (upper chamber) + MRL/lpr mice OBs (lower chamber); C+M+shRNA:C57/BL6 mice B cells (upper chamber) + MRL/lpr mice OBs transfected with shRNA (lower chamber); M+C: MRL/lpr mice B cells (upper chamber) + C57/BL6 mice OBs (lower chamber); M+M: MRL/lpr mice B cells (upper chamber) + MRL/lpr mice OBs (lower chamber). C: Number of C57-derived splenic B cells migrated to MRL/lpr-derived OBs treated with CXCL12 antagonists (LIT-927) at different concentration. D: Number of MRL/lpr-derived splenic B cells migrated to MRL/lpr-derived OBs treated with LIT-927 at different concentration. Data represented the mean± SD. *P<0.05, ** P<0.01, *** P<0.001.
Higher levels of CXCL12 enhanced the homing of B-lymphocytes to the marrow of MRL/lpr mice in vivo
Next, we performed an in vivo experiment to explore the mechanism of abnormal chemotaxis of B-lymphocytes in SLE mice. First, B cells were harvested and separated from the spleens of MRL/lpr mice and labeled these by CFDA. The labeled cells were then intravenously injected into recipient mice. After infusion, the presence of donor-derived B cells in the recipient BM was subsequently measured by flow cytometry. Among all three cohorts, the MRL/lpr cohort exhibited the highest percentage of CFDA-labeled cells. However, if the recipient mice were pretreated with LIT-927, the chemotactic capacity of infused B-lymphocytes to the BM was significantly impaired. In addition, no difference in the percentage of CFDA-labeled cells in the marrow was found between the C57 or MRL/lpr cohorts in the presence of LIT-927 (Figure 4). Taken together, these results once again indicated that marrow-derived CXCL12 is one of the key factors mediating enhanced B-lymphocyte homing to marrow.
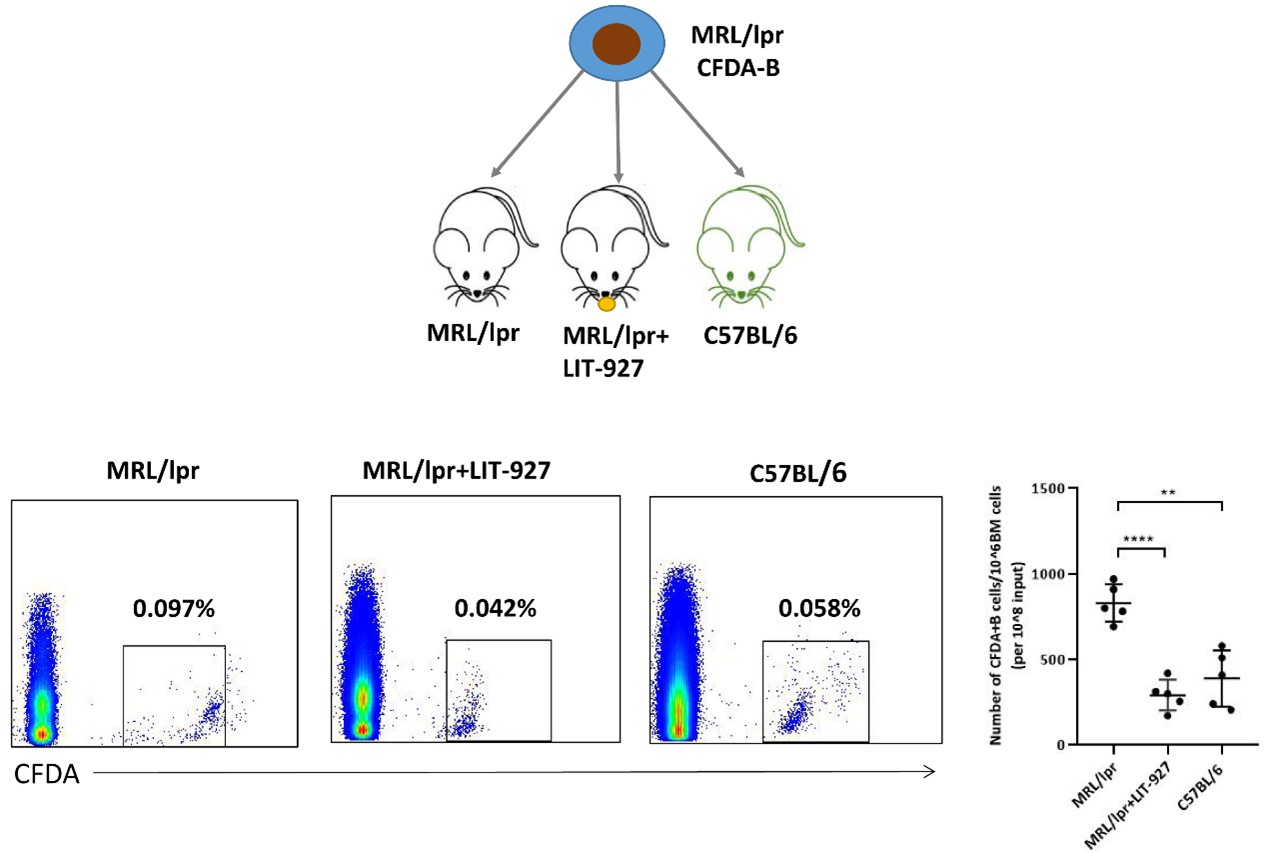
Figure 4. The number of CFDA-labeled MRL/lpr-derived B cells homing to BM. CFDA-labeled CD19+ B cells were retro-orbitally injected into 12- to 13-week-old syngenic C57BL/6 (n=5), MRL/lpr (n=5), and LIT-927 treated MRL/lpr (n=5), respectively. The percentage of CFDA-labeled B cells in the bone marrow was analyzed 24 hours later post-injection by flow cytometry. Data represented the mean ± SD. ** P<0.01, **** P<0.0001.
An increasing number of B lymphocytes preferentially colocalized with periosteal OBs in the BM of MRL/lpr mice
All the abovementioned results indicated that OBs overexpressing CXCL12 are more capable of attracting B-lymphocytes to their vicinity. To observe the spatial relationships between OBs and B-lymphocytes in vivo, we then immunostained femoral sections with antibodies against B220 (marker for B-lymphocytes), CD31 (marker for OBs), CD51 (marker for OBs) and CXCL12. Consistent with the flow cytometry results, more B220-positive B-lymphocytes were found in MRL/lpr mice, and the difference in the number of B-lymphocytes between MRL/lpr and C57 mice was more pronounced near the periosteal zone, where markedly fewer periosteal B-lymphocytes were found in C57 mice. We also found higher levels of CXCL12 expression in the MRL/lpr BM compared to C57 BM, which were consistent with the aforementioned research results. (Figure 5B). Interestingly, the CXCL12 gradient near the periosteal zone seemed to be more marked in MRL/lpr mice (Figure 5A). In addition, more CD51+CD31- OBs were present in the periosteal zone in MRL/lpr mice. Moreover, the colocalization of B220+ B lymphocytes and CD51+CD31- OBs could readily be observed in MRL/lpr mice (Figure 5C).
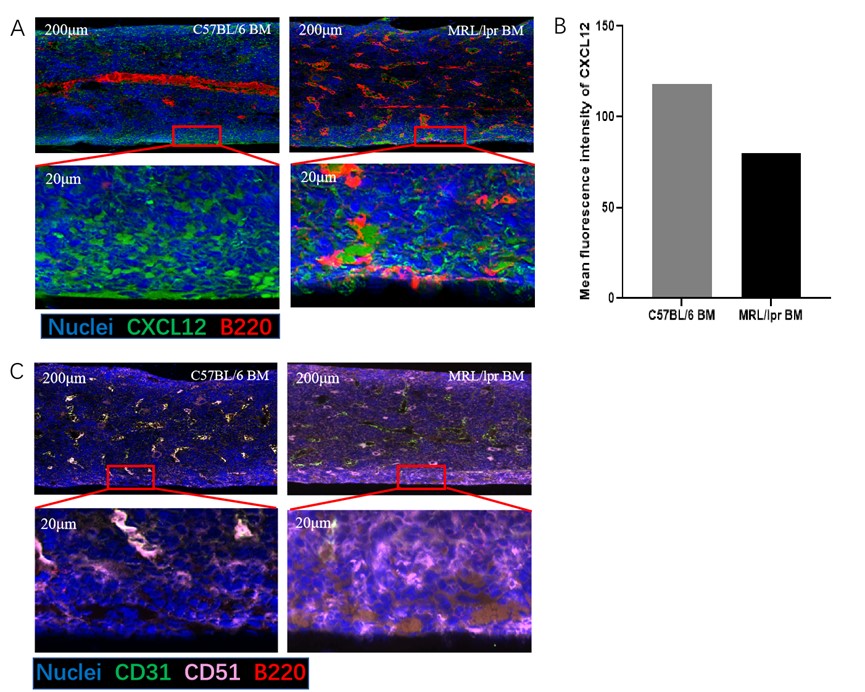
Figure 5. Representative image of C57 and MRL/lpr mice. Compared to C57mice, More B cells localize close to periosteal surface (A) and OB cells (B) where contains rich CXCL12 aggregates in MRL/lpr mice. Paraffin sections of mice femurs were assessed for the positivity and position of CXCL12, B cells (B220+ cells) and OB cells (CD31-CD51+) using immunofluorescence under confocal microscopy. The part in the box is magnified tenfold. Sections were counterstained with DAPI to visualize nuclei. A: Blue = DAPI, Red = B220, Green = CXCL12; B: Pink = CD51, Green = CD31, Red = B220. Scale bar = 200 μm. B: Differences between the expression of CXCL12 between C57 and MRL/lpr BM have been analyzed by Image J.
Correlation between percentages of CXCR4+ B cells or level of CXCL12 in BM and markers of disease activity in SLE patients
As shown in Figure 6, the percentages of CXCR4+ B cells were negatively correlated with the serum IgG concentration (r=-0.674, P<0.05). The level of CXCL12 was positively correlated with the systemic lupus erythematosus disease activity index (SLEDAI) score (r=0.745, P<0.05) and negatively correlated with the complement 3 (C3) concentration (r=-0.674, P<0.05). These clinical findings once again indicated that the abnormal BM environment characterized by increased levels of CXCR4+ B cells and CXCL12 is correlated with SLE disease activity.
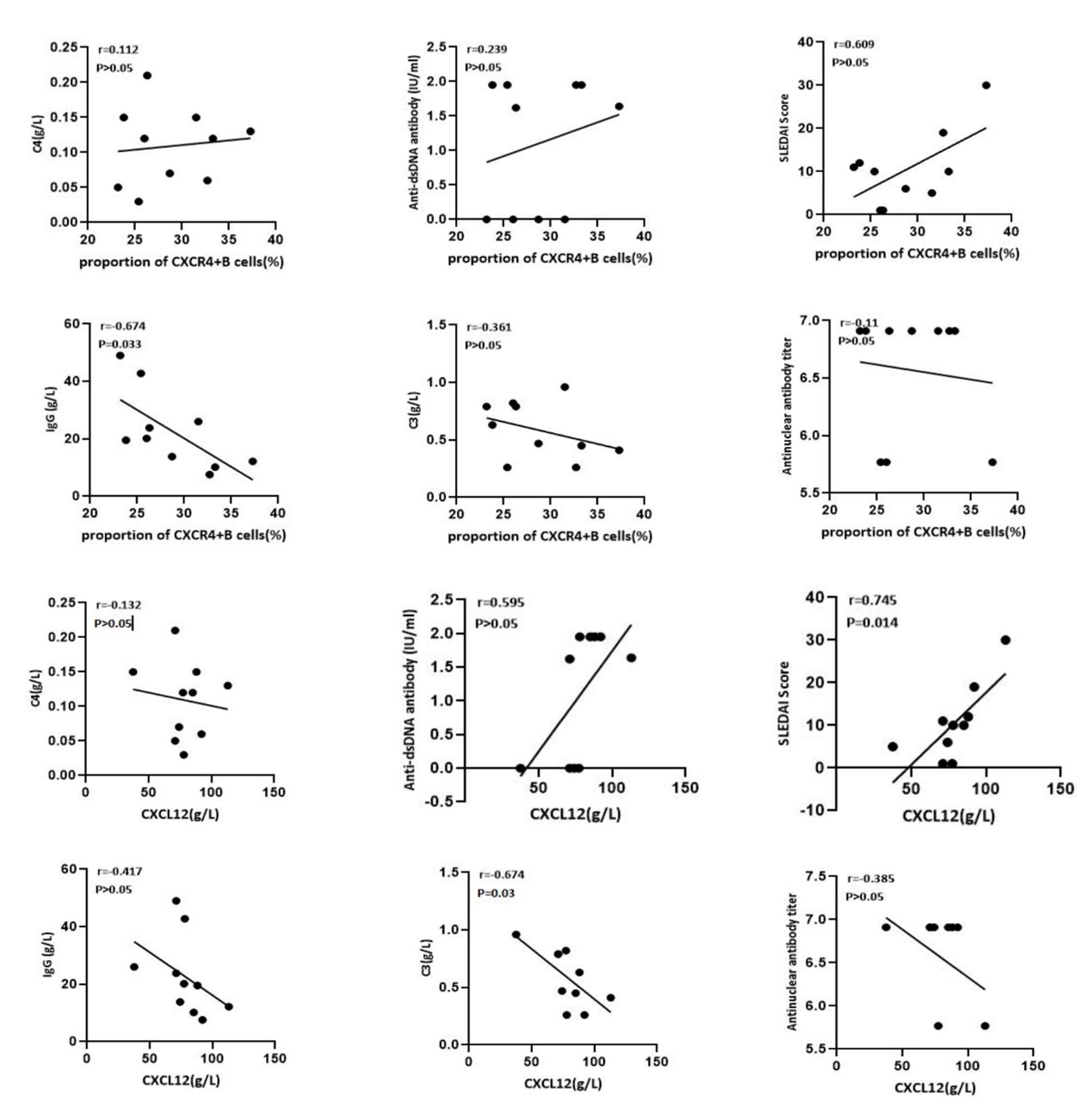
Figure 6. Both the percentages of CXCR4+ B cells and the level of CXCL12 in the bone marrow of SLE patients correlated with disease status. The percentages of CXCR4+ B cells were negatively correlated with IgG concentration (r=-0.674, P<0.05); The level of CXCL12 was positively correlated with SLEDAI Score (r=0.745, P<0.05) and negatively correlated with C3 concentration (r=-0.674, P<0.05).
Discussion
The key role of B-lymphocyte abnormalities in the pathogenesis of SLE has been widely accepted [1,2,5,24]. A standard view of B cells in systemic autoimmunity is that they promote lupus by producing autoantibodies. Upon activation, naïve B-lymphocytes differentiate into memory B cells and plasma cells (PCs), both of which are responsible for the production of pathogenic antibodies in SLE [24-26]. However, this view is incomplete because recent studies have revealed that autoimmune disease can be dissociated from auto-Ab deposition [2,24]. Therefore, B-cell-depleting therapy with anti-CD20 antibodies such as rituximab has been employed for the treatment of SLE in some clinical settings but with limited success [4,5]. Therefore, comprehensive elucidation of the mechanisms of self-reactive B-lymphocyte resistance to SLE treatment is important.
Previous studies have shown that the microenvironment not only is important for the development and survival of B-lymphocytes [27] but also provides a survival niche for memory-B cells and PC [26,28]. We previously found an abnormal BM microenvironment in SLE subjects [19,20], which is believed to be involved in the pathogenesis of B-lymphocyte abnormalities. The MRL/lpr mouse is one of most commonly used lupus models [29]. A decreased percentage of CXCR4+ B-lymphocytes is found in the PB of MRL/lpr mice compared with that of C57 control mice, and similar results can also be found in SLE patients. Theoretically, this observed decrease in CXCR4+ B-lymphocytes in the PB may result from either the exhaustion or redistribution of lymphocytes.
The CXCL12/CXCR4 axis reportedly mediates the chemotactic migration of multiple types of cells, such as neutrophils, monocytes, and B cells [6,8]. SDF-1 plays an important role in the accumulation of CXCR4+ immune cells in inflammatory tissues and chemoattraction of CXCR4+ tumor cells [30]. In NZB/W mice with nephritis manifestation, a high level of CXCL12 is hypothesized to be associated with increased renal inflammation and local autoantibody deposition due to inflammatory cell migration [31]. We found that B-lymphocytes from MRL/lpr mice showed increased migration along the CXCL12 gradient compared with those from C57 mice. Moreover, the mRNA level of CXCL12 was higher in the BM than in the PB and spleen (Figure 1C) in both C57 and MRL/lpr mice. These findings indicated that CXCR4+ B-lymphocytes preferentially migrate to the BM due to a higher CXCL12 gradient [14,15]. Furthermore, the level of CXCL12 in the BM was positively correlated with the SLEDAI score and anti-dsDNA antibody titer and negatively correlated with the serum C3 concentration. Based on these findings, we postulated that enhanced migration of CXCR4+ B cells to the BM might participate in the pathogenesis of SLE.
Many types of cells in the BM, including BMMSCs, vascular endothelial cells, and OBs, secrete CXCL12. In the current study, MRL/lpr-derived OBs were found to produce higher levels of CXCL12 than BMMSCs, which is one of many key factors for enhanced migration of B-lymphocytes both in vitro and in vivo. In addition to its chemotactic effect, the CXCL12/CXCR4 axis has also been demonstrated to be of paramount importance for B-cell lymphopoiesis and survival; for example, mice lacking CXCL12 die perinatally, and the numbers of B-cell progenitors in mutant embryos are severely reduced in the BM [6,15]. Reportedly, OBs can support many types of cells residing in the BM by secreting CXCL12 [32]: OBs can maintain the stemness of HSCs within marrow HSC niches [33]. When B cells colocalize with OBs, the differentiation of OBs may be blocked to prevent B-lymphocytes from losing their niches [34]. In the current study, more B220-positive B-lymphocytes were found in the periosteal zone of MRL/lpr mice compared with C57 mice, which was in accordance with the increased number of CD51+CD31- OBs found in the former. Moreover, B220-positive B lymphocytes were found to be spatially colocalized with CD51+CD31- OBs, where CXCL12-rich cells aggregated, and the colocalization of the two cellular components was more noticeable in MRL/lpr mice. These findings indicated that CXCL12-overexpressing OBs in MRL/lpr mice might play an important role in B-cell localization in the BM microenvironment.
After activation, B lymphocytes transform into immunoglobulin-producing immunoblasts and subsequently PCs. Long-lived PCs originate from proliferating plasma blasts in the spleen or lymph nodes, from where they migrate to the BM along a chemokine gradient [25]. After arriving in the BM, these cells must occupy a specific survival niche to mature into long-lived PCs. In addition, studies have also found that autoantibody-secreting PCs can be detected within inflamed kidneys in both SLE patients and murine lupus models [35-37]. Although the number of long-lived PCs in the BM decreases after target agents such as bortezomib [38], the residual cells are usually resistant to immunosuppressive agents and B-cell depletion therapy, and it is assumed that this resistance is the main cause of the refractoriness/relapse of autoimmune diseases [29,39]. Long-lived PCs preferentially dock with CXCL12-expressing stromal cells [40]. Dysregulated CXCL12-expressing OBs promote abnormal B-lymphomagenesis in SLE [15,17], and some cytokines and adhesion molecules, such as interleukin-6 (IL-6) and CXCL12, which are also expressed by stromal cells, provide long-lived PCs with additional survival signals [41,42]. Because high percentages of CXCR4+ B cells were found in the BM of SLE patients and MRL/lpr mice and were negatively correlated with the serum IgG levels in the PB, abnormal OBs in the BM might attract more PCs to the BM through the CXCL12/CXCR4 axis and further transform into long-lived PCs. Currently, immunotherapy for spine tumors has become a novel treatment, including checkpoint inhibitors, cancer vaccines, and chimeric antigen receptor T cells (CAR-T cells) [43,44]. Similarly, CXCL12 antagonist provides the potential for greater control of SLE. Nevertheless, the pathology of long-lived PCs in the BM in relapse/refractory SLE needs more investigation.
Conclusion
In summary, our study implies that the abnormal CXCR4/CXCL12 axis is associated with a shift in the distribution of B-lymphocytes between the BM and BM compartments in an SLE mouse model. More importantly, an increasing number of B-lymphocytes were found to colocalize with CXCL12-expressing OBs in the periosteal zone. We hypothesize that dysregulated OB niches play an important role in the pathogenesis of SLE, and the clinical significance of this finding deserves further exploration.
Supplementary Material
The demographic information of the SLE patients and healthy controls is shown in Supplementary Table S1.
No significant difference in age was found between the two groups. Clinical information of SLE patients is shown in Supplementary Table S2.
BM cells from C57BL/6 and MRL/lpr mice (the methods are described in ‘BM mesenchymal stem cell culture’) were used for the colony formation assay. The number of CFU-pre-B was lower in MRL/lpr mice than in C57 mice (n=5, P <0.05) (Figure S3).
Data Availability
The data (figures and table) used to support the findings of this study are included within the article.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Authors’ Contributions
WZ, CM, and MC participated in the conception and design of the study; WZ and WS performed the analysis and interpretation of the data; WZ drafted the manuscript; YT and XF reviewed the manuscript. All the authors read and approved the manuscript.
Acknowledgments
We thank Dr. Zhongqun Wang for his kind help with this study.
Sources of Funding
This work was supported by the Natural Science Foundation of China (81571582) and Society Development Foundation of Jiangsu Province-Advanced Clinical Technology (BE2020681).
Ethics Approval
All experiments with human samples were approved by the Ethics Committee of the Affiliated Hospital of Jiangsu University (reference number: KY2021K0909). All animal experiments were approved by the Animal Ethics Committee of the Jiangsu University Animal Laboratory Center (reference number: UJS-IACUC-AP-2020032539).
References
2. Chan OT, Madaio MP, Shlomchik MJ. The central and multiple roles of B cells in lupus pathogenesis. Immunol Rev. 1999;169:107-21.
3. Sayyed SG, Hägele H, Kulkarni OP, Endlich K, Segerer S, Eulberg D, et al. Podocytes produce homeostatic chemokine stromal cell-derived factor-1/CXCL12, which contributes to glomerulosclerosis, podocyte loss and albuminuria in a mouse model of type 2 diabetes. Diabetologia. 2009;52(11):2445-54.
4. Smith KG, Jones RB, Burns SM, Jayne DR. Long-term comparison of rituximab treatment for refractory systemic lupus erythematosus and vasculitis: Remission, relapse, and re-treatment. Arthritis Rheum. 2006;54(9):2970-82.
5. Chan VS, Tsang HH, Tam RC, Lu L, Lau CS. B-cell-targeted therapies in systemic lupus erythematosus. Cell Mol Immunol. 2013;10(2):133-42.
6. Wang A, Fairhurst AM, Tus K, Subramanian S, Liu Y, Lin F, et al. CXCR4/CXCL12 hyperexpression plays a pivotal role in the pathogenesis of lupus. J Immunol. 2009;182(7):4448-58.
7. Chen G, Chen SM, Wang X, Ding XF, Ding J, Meng LH. Inhibition of chemokine (CXC motif) ligand 12/chemokine (CXC motif) receptor 4 axis (CXCL12/CXCR4)-mediated cell migration by targeting mammalian target of rapamycin (mTOR) pathway in human gastric carcinoma cells. J Biol Chem. 2012;287(15):12132-41.
8. Yi L, Zhou X, Li T, Liu P, Hai L, Tong L, et al. Notch1 signaling pathway promotes invasion, self-renewal and growth of glioma initiating cells via modulating chemokine system CXCL12/CXCR4. J Exp Clin Cancer Res. 2019;38(1):339.
9. Hanaoka H, Okazaki Y, Hashiguchi A, Yasuoka H, Takeuchi T, Kuwana M. Overexpression of CXCR4 on circulating B cells in patients with active systemic lupus erythematosus. Clin Exp Rheumatol. 2015;33(6):863-70.
10. Barrera-Vargas A, Gómez-Martín D, Carmona-Rivera C, Merayo-Chalico J, Torres-Ruiz J, Manna Z, et al. Differential ubiquitination in NETs regulates macrophage responses in systemic lupus erythematosus. Ann Rheum Dis. 2018;77(6):944-50.
11. de la Varga Martínez R, Rodríguez-Bayona B, Añez GA, Medina Varo F, Pérez Venegas JJ, Brieva JA, et al. Clinical relevance of circulating anti-ENA and anti-dsDNA secreting cells from SLE patients and their dependence on STAT-3 activation. Eur J Immunol. 2017;47(7):1211-9.
12. Balabanian K, Couderc J, Bouchet-Delbos L, Amara A, Berrebi D, Foussat A, et al. Role of the chemokine stromal cell-derived factor 1 in autoantibody production and nephritis in murine lupus. J Immunol. 2003;170(6):3392-400.
13. Cheng Q, Khodadadi L, Taddeo A, Klotsche J, B FH, Radbruch A, et al. CXCR4-CXCL12 interaction is important for plasma cell homing and survival in NZB/W mice. Eur J Immunol. 2018;48(6):1020-9.
14. Power CA. Knock out models to dissect chemokine receptor function in vivo. J Immunol Methods. 2003;273(1-2):73-82.
15. Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, et al. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 1996;382(6592):635-8.
16. Allen CD, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. 2004;5(9):943-52.
17. Ma Q, Jones D, Borghesani PR, Segal RA, Nagasawa T, Kishimoto T, et al. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc Natl Acad Sci U S A. 1998;95(16):9448-53.
18. Janssens R, Struyf S, Proost P. Pathological roles of the homeostatic chemokine CXCL12. Cytokine Growth Factor Rev. 2018;44:51-68.
19. Tang Y, Xie H, Chen J, Geng L, Chen H, Li X, et al. Activated NF-kB in bone marrow mesenchymal stem cells from systemic lupus erythematosus patients inhibits osteogenic differentiation through downregulating Smad signaling. Stem Cells Dev. 2013;22(4):668-78.
20. Tang Y, Ma X, Zhang H, Gu Z, Hou Y, Gilkeson GS, et al. Gene expression profile reveals abnormalities of multiple signaling pathways in mesenchymal stem cell derived from patients with systemic lupus erythematosus. Clin Dev Immunol. 2012;2012:826182.
21. Visnjic D, Kalajzic Z, Rowe DW, Katavic V, Lorenzo J, Aguila HL. Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood. 2004;103(9):3258-64.
22. Aronow MA, Gerstenfeld LC, Owen TA, Tassinari MS, Stein GS, Lian JB. Factors that promote progressive development of the osteoblast phenotype in cultured fetal rat calvaria cells. J Cell Physiol. 1990;143(2):213-21.
23. Semerad CL, Christopher MJ, Liu F, Short B, Simmons PJ, Winkler I, et al. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood. 2005;106(9):3020-7.
24. Dörner T, Giesecke C, Lipsky PE. Mechanisms of B cell autoimmunity in SLE. Arthritis Res Ther. 2011;13(5):243.
25. Dörner T, Radbruch A, Burmester GR. B-cell-directed therapies for autoimmune disease. Nat Rev Rheumatol. 2009;5(8):433-41.
26. Malkiel S, Barlev AN, Atisha-Fregoso Y, Suurmond J, Diamond B. Plasma Cell Differentiation Pathways in Systemic Lupus Erythematosus. Front Immunol. 2018;9:427.
27. Zhu J, Garrett R, Jung Y, Zhang Y, Kim N, Wang J, et al. Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood. 2007;109(9):3706-12.
28. Balakumaran A, Robey PG, Fedarko N, Landgren O. Bone marrow microenvironment in myelomagenesis: its potential role in early diagnosis. Expert Rev Mol Diagn. 2010;10(4):465-80.
29. Crampton SP, Morawski PA, Bolland S. Linking susceptibility genes and pathogenesis mechanisms using mouse models of systemic lupus erythematosus. Dis Model Mech. 2014;7(9):1033-46.
30. Kucia M, Jankowski K, Reca R, Wysoczynski M, Bandura L, Allendorf DJ, et al. CXCR4-SDF-1 signalling, locomotion, chemotaxis and adhesion. J Mol Histol. 2004;35(3):233-45.
31. Devarapu SK, Kumar Vr S, Rupanagudi KV, Kulkarni OP, Eulberg D, Klussmann S, et al. Dual blockade of the pro-inflammatory chemokine CCL2 and the homeostatic chemokine CXCL12 is as effective as high dose cyclophosphamide in murine proliferative lupus nephritis. Clin Immunol. 2016;169:139-47.
32. Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, et al. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. 2010;33(3):387-99.
33. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25(6):977-88.
34. Sun W, Meednu N, Rosenberg A, Rangel-Moreno J, Wang V, Glanzman J, et al. B cells inhibit bone formation in rheumatoid arthritis by suppressing osteoblast differentiation. Nat Commun. 2018;9(1):5127.
35. Espeli M, Bökers S, Giannico G, Dickinson HA, Bardsley V, Fogo AB, et al. Local renal autoantibody production in lupus nephritis. J Am Soc Nephrol. 2011;22(2):296-305.
36. Sekine H, Watanabe H, Gilkeson GS. Enrichment of anti-glomerular antigen antibody-producing cells in the kidneys of MRL/MpJ-Fas(lpr) mice. J Immunol. 2004;172(6):3913-21.
37. Starke C, Frey S, Wellmann U, Urbonaviciute V, Herrmann M, Amann K, et al. High frequency of autoantibody-secreting cells and long-lived plasma cells within inflamed kidneys of NZB/W F1 lupus mice. Eur J Immunol. 2011;41(7):2107-12.
38. Alexander T, Cheng Q, Klotsche J, Khodadadi L, Waka A, Biesen R, et al. Proteasome inhibition with bortezomib induces a therapeutically relevant depletion of plasma cells in SLE but does not target their precursors. Eur J Immunol. 2018;48(9):1573-9.
39. Shachar I, Karin N. The dual roles of inflammatory cytokines and chemokines in the regulation of autoimmune diseases and their clinical implications. J Leukoc Biol. 2013;93(1):51-61.
40. Moser K, Tokoyoda K, Radbruch A, MacLennan I, Manz RA. Stromal niches, plasma cell differentiation and survival. Curr Opin Immunol. 2006;18(3):265-70.
41. Cassese G, Arce S, Hauser AE, Lehnert K, Moewes B, Mostarac M, et al. Plasma cell survival is mediated by synergistic effects of cytokines and adhesion-dependent signals. J Immunol. 2003;171(4):1684-90.
42. Minges Wols HA, Underhill GH, Kansas GS, Witte PL. The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J Immunol. 2002;169(8):4213-21.
43. Chalamgari A, Valle D, Palau Villarreal X, Foreman M, Liu A, Patel A, et al. Vertebral Primary Bone Lesions: Review of Management Options. Curr Oncol. 2023;30(3):3064-78.
44. Amadasu E, Panther E, Lucke-Wold B. Characterization and Treatment of Spinal Tumors. Intensive Care Res. 2022;2(3-4):76-95.