Abstract
Introduction: Neonatal seizures are associated with neurodevelopmental impairments. Implementing long-term video-EEG monitoring in the neonatal intensive care unit became the gold standard for seizure diagnosis. During the neonatal period, seizures can be associated with an acute brain insult called acute symptomatic seizures (ASS) or being part of neonatal epilepsy that may have a structural, metabolic, or genetic cause. This distinction impacts patient workup and management. Objectives: To facilitate a guide to differentiate ASS from neonatal epilepsy, and to correlate different electroclinical seizure patterns with a specific etiology. Methods: A narrative review was performed. MEDLINE, Embase, and PubMed were used to gather data for this narrative review. The following keywords were applied to focus on original research and case reports: epileptic encephalopathy, developmental Epileptic encephalopathy and neonatal seizures, neonatal genetic encephalopathies, Otahara syndrome, neonatal channelopathies, and neonatal seizure classification. Conclusions: Strict electroclinical semiology is the backbone for diagnosing neonatal seizures. The EEG and ictal semiology help with the diagnosis and the treatment. The neonatal seizure classification should be expanded to include the EEG pattern. Lumping them in a better classificatory system will prevent unnecessary and hazardous medication.
Keywords
Epileptic encephalopathy, Neonatal seizures, Burst-Suppression pattern, Channelopathy, Acute symptomatic seizures, Antiseizure medication
Introduction
Seizures in the neonatal period (birth to the first 28 postnatal days or up to 44 weeks postconceptional age) affect 1–3 per 1000 births and are associated with neurodevelopmental impairments [1]. Implementing long-term video-EEG monitoring in neonatal intensive care units has become the gold standard for seizure diagnosis, characterization, and accurate quantifying seizure burden in newborns [2]. Neuroimaging and metabolic and genetic testing have played an essential role in identifying the underlying causes of seizures in neonates [3]. During the neonatal period, seizures can be associated with either: acute brain insults called acute symptomatic seizures (ASS) [4] or neonatal epilepsy that may have a structural, metabolic, or genetic cause [5].
It has been recognized that seizure semiology and particular electroencephalogram patterns provide clues about the underlying cause of epilepsy, offering a diagnostic value with subsequent treatment implications [3,6-8].
As a result, the ILAE Commission on Classification and Terminology accepts that seizures in neonates require special attention; therefore, a Neonatal Task Force integrates neonatal seizures and epilepsies into the 2017 ILAE Classification [8]. However, the current classification system is still incomplete and grouped under the same category (i.e.) "structural etiology" different causes with different electroclinical features and treatments [8].
This communication aims to facilitate a guide to differentiate ASS from neonatal epilepsy and to correlate different electroclinical seizure patterns with a specific etiology, with subsequent impact on treatment decisions.
Electroclinical Approach
This electroclinical approach is based on a literature review and is not intended to substitute clinical judgment. Medical history, prenatal history, perinatal history, and physical examination are the backbone for diagnosis in medicine.
Neonatal seizures are always focal and can be electroclinical or electrographic in nature. The diagnosis cannot be made acutely without an electroencephalogram correlation. Electrographic seizures are hallmarks of ASS. The etiology and seizure burden dictate prognosis [9].
Acute symptomatic seizures vs. neonatal epilepsy
Acute symptomatic seizures: In the neonatal period, ASS presents with electrographic seizures (Figure 1) or unilateral focal clonic seizures. Unilateral clonic seizures are the most frequent presentation of ischemic stroke in the neonatal period [5]. Most often, these are the only clinical signs. Clonic seizures can be multifocal and mainly represent the herald of neonatal infection, reflecting a diffuse acute brain insult. ASS is rarely presented with autonomic seizures [5,9,10]. However, apneic seizures are observed in intraventricular or intraparenchymal hemorrhagic strokes, particularly affecting the temporal lobe [12].
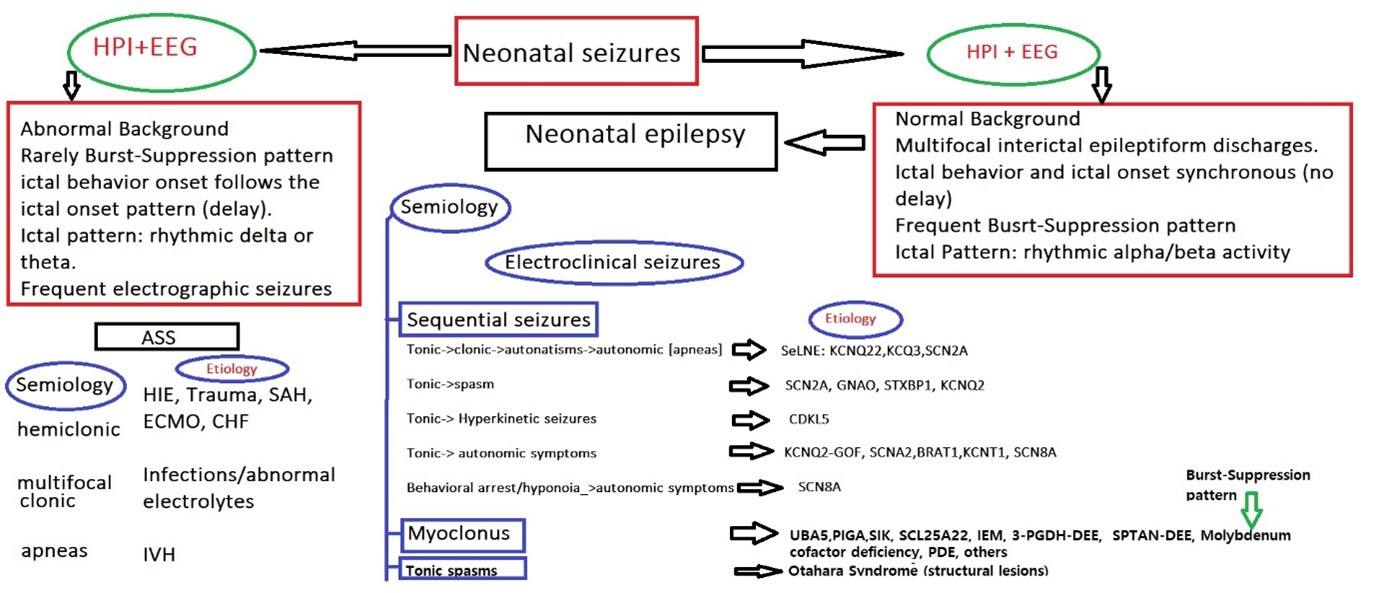
Figure 1. Proposed neonatal seizures classification and clinical approach. KCNQ22 (K” for the scientific symbol for potassium, “CN” as an abbreviation for channel, and “Q2” for the subtype Q2), KCNQ3 (Q3 subtype), SCN2A ( sodium or salt channel), SeLNE (Self-limited neonatal Epilepsy), KCNQ2 (K” for the scientific symbol for potassium, “CN” as an abbreviation for channel, and “Q2” for the subtype Q2), STXBP1(Syntaxin-binding protein 1 ), GNAO1(G Protein Subunit Alpha O1), CDKL-5 (cyclin-dependent kinase-like 5), UBA5 (the ubiquitin-activating enzyme of UFM1.), PIGA (Phosphatidylinositol N-acetylglucosaminyltransferase subunit A (PIG-A), SIK (salt-inducible kinase (SIK), one of the AMP-activated kinase (AMPK)-related kinases), SCL25A2 (solute carrier family 5 member 2), IEM (Inborn Error of Metabolism), HF (heart failure), Extracorporeal membrane oxygenation (ECMO), HIE (hypoxic Ischemic Encephalopathy), SAH (subarachnoid hemorrhage), IVH (intraventricular hemorrhage), HPI (history of present illness), ASS (Acute Symptomatic Seizures.
Electrographic seizures are common in HIE, neonates with congenital diaphragmatic hernia and heart disease, those exposed to extracorporeal membrane oxygenation, or while being sedated and paralyzed [12].
ASS usually subsides after an acute event, and there is no need to continue ASM treatment after discharge [9].
Most of the time, the electroencephalogram (EEG) in ASS shows pathological discontinuity, focal slowing, asymmetries in frequency and amplitude, and multifocal spikes. However, a clear, persistent epileptiform focus is occasionally observed. Seizures can originate from only one brain area or can be observed multifocally, but electrographic seizures never appear to change sequentially from one hemisphere to another (Figure 2A). Seizures in neonates with ASS appear after a time-lapse interval following the onset of the ictal pattern on the EEG. The ictal pattern usually shows a focal, slowly evolving rhythm, and the postictal period does not show prolonged postictal depression [2,6,13].
Neonatal epileptic seizures: Neonatal epilepsies have a completely different presentation and can be suspected from clinical and electroencephalographic points of view (Figure 1). Neonates with epileptic channelopathies or synaptopathies can be recognized as presenting with short tonic or sequential seizures with an initial tonic component. Different seizure sequences can be recognized [11,12,14]:
- Tonic followed by clonic and autonomic symptoms typically seen in KNCQ2-Developmental Epilepsy Encephalopathy (DEE) and SNC2A-DEE.
- Tonic followed by epileptic spasm (typically observed in STXBP1-DDE).
- Tonic seizures followed by hyperkinetic movements are observed in CDKL-5-DEE.
- Tonic followed by autonomic features (cyanosis or apneas) seen in SCN2A-DEE, KCNT1-DEE, SCN8A-DEE, KCNQ2-GOF-DEE, BRAT1 -DEE or behavioral arrest due to hypotonia followed by autonomic symptoms seen in SCN8A-DEE.
- Early infantile DEE with myoclonus is seen in Inborn Error of Metabolism (IEM) or in SIK-DEE, UBA5-DEE, PIGA-DEE, SIK-DEE, and SCL25A2-DEE [4,15,16] (Figure 1).
Neonatal seizures are associated with typical ictal and interictal EEG patterns. Unlike ASS, the EEGs show normal background activity associated with focal or multifocal spikes and wave discharges [11,17-19]. Myoclonic seizures are associated with burst–suppression patterns, and two epileptic syndromes can be distinguished: Otahara Syndrome (OS), characterized by tonic spasms (spasms lasting up to 5 seconds), and Burst Suppression pattern during sleep and wake state. Early Infantile Myoclonic Epileptic Encephalopathy has seizures consisting of myoclonic, clonic, and spasms, and burst-suppression patterns that cannot be seen during the awake state [4,15,16,20,21]. OS is associated with structural abnormalities (malformation of cortical development (MCD), while EIMEE (Early Infantile Myoclonic Epileptic Encephalopathy) is associated with IEM [21]. Spasms in OS can be asymmetric, symmetric, or unilateral. Typically, asymmetric tonic spasms are observed in neonates with significant brain malformations, such as hemimegaloencephaly [22-24].
The ictal EEG patterns in neonatal channelopathies consist of fast activity with rapid spread to the adjacent and contralateral areas of the head. Prolonged postictal depression characterizes the postictal period. From an EEG point of view, neonates with genetic epilepsies present with brief seizures that are evident from the onset of electrographic seizures. They are synchronous, and there is no time lapse between ictal EEG onset and behavioral changes, as seen in ASS [1-3,6].
Neonatal epilepsies (Figure 1)
ILAE recognizes three neonatal epilepsies [8,14].
1. Self-limited Neonatal Epilepsy (SeLNE).
2. Otahara syndrome (OS).
3. Early Myoclonic Encephalopathy (EME).
Self-limited neonatal epilepsy: In most cases, SeLNE is associated with normal development. Sequential tonic seizures characterize its seizures. The ictal sequence with tonic seizure followed by clonic jerks and hand or leg automatisms evolving to non-motor autonomic features, such as apnea, is commonly seen. The EEG shows a normal background. Seizures can start in one hemisphere but change to the other, and the postictal period shows diffuse attenuation [1,3,14,18].
KCNQ2-related Epilepsy: This gene's loss-of-function (LoF) variants were first linked to SeLNE in 1998 and were also found to be responsible for neonatal-onset DEE in 2012. SeLNE and KCNQ2-DEE are part of a large spectrum, where SeLNE is the less severe phenotype, and DEE is the most severe. In KCNQ2-SeLNE, infants present at 2–5 days of life with isolated or multiple seizures per day, which may evolve into status epilepticus. Between seizures, neonates are healthy and can be breastfed unless phenobarbital is administered [18,25-27].
The EEG background is well organized, sometimes with focal epileptiform abnormalities during the peri-ictal phase (Figure 2B). Isolated seizures may resolve spontaneously within a few months; however, most neonates require ASM treatment. Later in life, they have normal motor and cognitive outcomes [28,29].
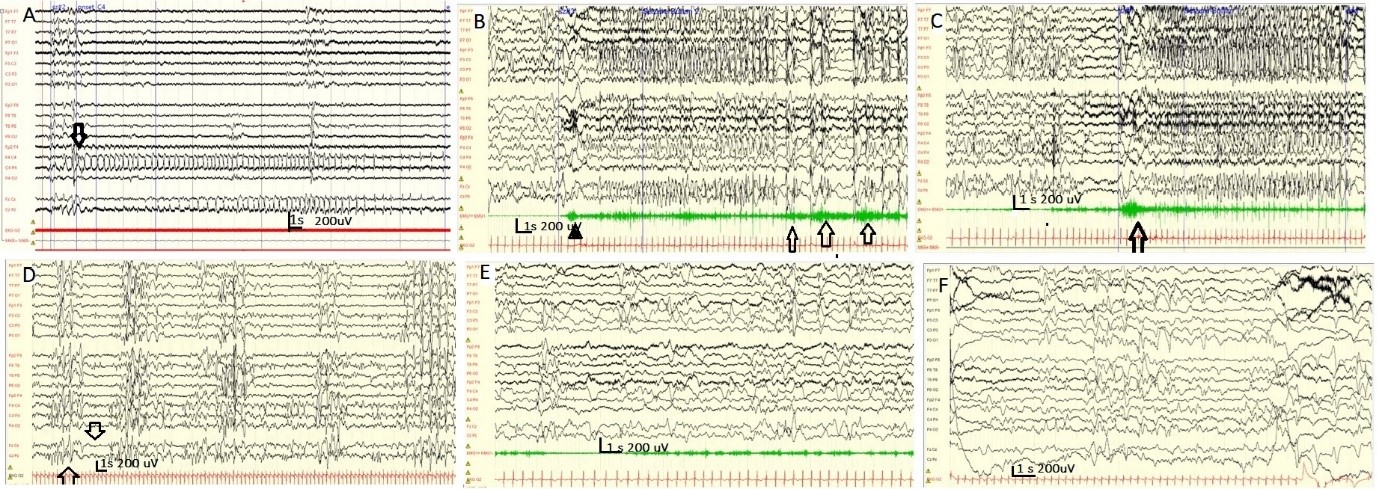
Figure 2. Different interictal and ictal patterns in ASS and neonatal epilepsy. A) Discontinuous background and focal electrographic seizures with C4 onset (see arrow) in a 40-week patient with HIE. B) Synchronous onset of high-amplitude slow waves followed by generalized rhythmic alpha activity associated with an epileptic spasm (arrow) in a patient with STXBP1-DEE. C) Sequential seizure spasm followed by brief tonic seizures ending with multifocal clonic jerks. Initially, a brief diamond-shaped myogenic artifact (arrowhead) was observed, followed by myoclonic and clonic jerks (see arrows) in a patient with KCNQ2-DEE. D, E, and F show the Burst -Suppression pattern during sleep (E, see arrows) and multifocal epileptiform discharges (D, F) and disappearance of the BS pattern in a patient with early onset Myoclonic Epileptic Encephalopathy.
DEE presenting with sequential seizures:
Sequential seizures or exclusively tonic seizures [11,12,14,30]:
- GNAO1-DEE
- KCNQ2- DEE
- STXBP1- DEE
- CDKL5- DEE
- SCN2A- DEE
Tonic seizures, rarely sequential seizures (isolate tonic seizures) (Figure 1): GNAO1-DEE most frequently presents as focal tonic seizures and hypotonia. The EEG background is abnormal, with either a suppression-burst pattern, hypsarrhythmia, or multifocal sharp waves. All infants experience profound developmental delays and movement disorders that appear later in life [31,32].
KCNQ2- DEE caused by pathogenic variants of the KCNQ2 gene, is presented with sequential or tonic seizures associated with a burst suppression pattern or multifocal spikes in the EEG. Patients with KCNQ2-DEE may have a family member with SeLNE [24,28,33-36].
Sequential seizures and asymmetric tonic seizures and spasms: STXBP1- DEE usually starts after the first month of life, but neonatal seizures have been described. The typical phenotype consists of early-onset epileptic spasms. EEG findings range from multifocal epileptiform abnormalities to suppression burst patterns. Recurrent MRI findings included a thin corpus callosum and frontal hypoplasia. STXBP1-DEE is associated with asymmetric tonic posturing or sequential seizures (tonic, autonomic, clonic, and epileptic spasms) (Figure 2C). Several studies have documented excellent responses to levetiracetam [7,27,29,37-40].
Sequential seizures typically hyperkinetic-tonic-spasms: CDKL5- DEE is associated with tonic seizures. Patients with CDKL-5-DEE have seizure onset between 1 week of age and 1.5 years. Usually, they have two seizures per day. Seizure semiology is characterized first by spasms. A peculiar pattern consisting of prolonged tonic and clonic events with a vibratory contraction, followed by a clonic phase and a cluster of spasms ending with myoclonus. EEG can be normal in the early stage, but it is followed by multifocal spikes and waves that evolve into hypsarrhythmia. Ictal EEG is characterized by generalized flattening, followed by repetitive sharp spikes and wave discharges. Sequential seizures typically evolve to a "hyperkinetic- tonic-spasms" phenotype during infancy [26,29,41].
Sequential seizures with tonic followed by autonomic features: SCN2A-DEE includes sequential seizures with predominantly tonic and autonomic features. Seizures usually begin in the first week of life. Seizure semiology consists of alternating focal clonic and tonic seizures, apnea, and cyanosis. Seizures are frequent, up to 100/day. The EEG shows an encephalopathic pattern characterized by a discontinuity that evolves into multifocal spikes. Improvement can be observed with sodium channel blockers (phenytoin) [19,25,42,43].
- KCNT1- DEE presents with focal tonic seizures and autonomic symptoms [18,44-46].
- BRAT1-DEE is an autosomal recessive disorder characterized by hypertonia, arthrogryposis, and spontaneous, nonepileptic, and multifocal myoclonus over a relatively organized EEG background. Neonates with BRAT1 encephalopathy develop intractable multifocal tonic seizures, followed by apneas and bradycardia, leading to early death. The accompanying symptoms are pivotal in suspecting a diagnosis (microcephaly and arthrogryposis) [18,47].
- Neonates with SCN1A GoF-DEE present in the very first days of life and develop hyperkinetic movement disorders with choreoathetosis later in life [18].
- KCNQ2-DEE represents a severe phenotype in the KCNQ2-related epilepsy spectrum. Neonates present shortly after birth with persistent seizures, up to 35 seizures per hour, and are encephalopathic. Their EEG background is abnormal, with multifocal epileptiform discharges and random attenuation or a suppression-burst pattern. Seizure semiology is characterized by focal sequential seizures with alternating laterality, consisting of asymmetric tonic posturing accompanied by apnea and desaturation, which evolve into unilateral or asynchronous bilateral clonic components [28,35]. These patients did not exhibit a cluster of spasms. Although typically seen in KCNQ2, this electroclinical phenotype can be seen in the other two known DEEs: KCNQ3-DEE and SCN2A-DEE [18,25,48]. There are patients with KCNQ2-GoF (gain of function) variants. Interestingly, these neonates do not have seizures in the neonatal period but suffer from stimulus-sensitive nonepileptic myoclonus, severe encephalopathy, central hypoventilation, and a suppression-burst pattern on EEG [34,35].
- Epilepsy caused by SCN2A GoF variants may present with seizures during early infancy with an excellent response to sodium channel blockers (SCBs). In contrast, patients with LoF variants tend to have later onset and poor response to SCBs [42,43].
DEE presenting with hypotonic seizures plus autonomic features: SCN8A- DEE: In this encephalopathy, seizures occur in clusters and are commonly observed during sleep. Prolonged focal seizures with prominent inhibitory components (hypomotor) associated with apnea, brady/tachycardia, and cyanosis evolving into unilateral tonic or clonic manifestations, and lately, bilateral tonic and clonic seizures are seen (Figure 3A). Non-convulsive status epilepticus has also been frequently described. Electroencephalograms (EEG) can be normal or mildly abnormal at epilepsy onset but evolve to a progressive background deterioration with interictal epileptiform discharges that predominate in the posterior regions of the head. The focal seizures have temporo-occipital EEG onset and might have migrated from one hemisphere to another. Patients with pathogenic variants in the SCN8A gene show seizure responses to sodium channel agents, often at high doses [42,43].
DEE presents with predominant myoclonic seizures: This group presents with a phenotype termed Early Infantile Onset Myoclonic Epileptic Encephalopathy (EIMEE). These patients had erratic, multifocal, and fragmentary epileptic myoclonus, usually in the context of IEM. Focal seizures, including eye deviation, asymmetric tonic posturing, facial flushing, and apnea, are also prevalent. Isolates or clusters of tonic spasms would be present. The EEG shows a burst-suppression pattern during sleep, disappearing during wakefulness (Figure 2D) [22,29,49]. One study reported a burst-suppression pattern only during sleep in 33% of cases. Another peculiar characteristic of this syndrome is that the Burst Suppression pattern may not be evident at epilepsy onset. Myoclonic movements are not associated with electrographic changes. The suppression burst pattern can evolve into an atypical hypsarrhythmia pattern [15,22].
EIMEE is an umbrella term for many channelopathies and IEM, such as D-glyceric acidemia, propionic aciduria, molybdenum cofactor deficiency, pyridoxine deficiency, methylmalonic acidemia, sulfite oxidase deficiency, Menkes disease, and Zellweger syndrome. Within this group of channelopathies, we found the following [50-56].
- UBA5-DEE [55,56].
- PIGA-DEE [55,56].
- SIK1-DEE [57].
- SLC25A22-DEE [29,46].
UBA5- DEE: UBA5- DEE is an autosomal recessive disorder characterized by early onset encephalopathy, movement abnormalities, global developmental delay, intellectual disability, and seizures. Seizure semiology is dominated by myoclonic jerk, although epileptic spasms can be seen [58].
PIGA-DEE: Phosphatidylinositol glycan biosynthesis class A protein germline mutations result in this severe form of early onset epileptic encephalopathy. Most of the seizures in this patient are characterized by myoclonus. The EEG reveals a suppression-burst pattern. Brain MRI showed a thin corpus callosum, delayed myelination, hypoplastic cerebellum, cortical atrophy, and restricted water diffusion in the brainstem, basal ganglia, and cerebellum. Most of the patients are hypotonic and have facial dysmorphism. Some patients have heart, liver, and kidney anomalies, as well as deafness and visual impairment. Serum alkaline phosphatase levels are elevated in most patients [27,59,60].
SIK1-DEE: The neonatal onset of early myoclonic seizures and tonic spasms characterizes SIK1-DEE. Patients with neonatal-onset SIK1-DEE have shorter survival. The EEG shows a burst suppression pattern. Although a specific treatment is not available, recognizing this epileptic encephalopathy would help decide the transition to conform to care [57].
SLC25A22- DEE: SLC25A22- DEE is characterized by a very early onset of epilepsy, usually occurring in the first two days of life. It is caused by a homozygous truncating mutation in the gene encoding the glutamic acid carrier. Epilepsy is severe. Myoclonic jerks characterize seizures, and the EEGs show a burst-suppression pattern. Patients usually have microcephaly, and the response to anti-seizure medication is never obtained, but phenobarbital can decrease the seizure burden. The visual-evoked potential is abnormal in these patients [29,46,61,62].
Pyridoxine-dependent epilepsy (PDE)-DEE: (PDE)-DEE is a rare, treatable form of metabolic epilepsy. Pathogenic variants in the ALDH7A1 gene affect cerebral lysine catabolism. Neonates have a very recognizable phenotype characterized by frequent focal and multifocal clonic and myoclonic seizures, which are refractory to common ASMs; as additional signs, infants usually present with marked irritability, fluctuating tone, and emesis. The specific EEG pattern may vary from normal to focal abnormalities to suppression-burst or hypsarrhythmia [22,55].
SLC13A5-DEE: SLC13A5-DEE appears during the first few days of life. SCL13A5 protein is a sodium-dependent citrate co-transporter. Neonatal epilepsy in this syndrome is characterized by intractable focal seizures that evolve into focal status epilepticus. These neonates predominantly have myoclonic and clonic seizures. The neurological examination reveals diffuse hypotonia. The EEG background is normal in most of these neonates; however, they may rarely show a suppression-burst pattern [63,64].
SPTAN-DEE: SPTAN-DEE appears during the neonatal period or infancy. The seizures consist of spasms in the first three weeks of life or rarely later. Usually, patients suffer from myoclonic or asymmetric tonic seizures. These seizures are refractory to antiepileptic and hormonal therapies. The EEGs show multifocal spikes, hypsarrhythmia, or modified hypsarrhythmia. The burst suppression pattern is not observed. Magnetic resonance imaging (MRI) shows pontocerebellar atrophy [62,65].
3-phosphoglycerate dehydrogenase deficiency-DEE: The clinical manifestations of 3-phosphoglycerate dehydrogenase deficiency is characterized by congenital microcephaly, profound mental retardation, hypertonia, intractable seizures, and occasional West Syndrome. 3-Phosphoglycerate dehydrogenase deficiency is diagnosed by amino acid analysis of the plasma and cerebrospinal fluid. A diagnosis is made by analyzing CSF amino acid levels, with the characteristic abnormalities best appreciated in the fasting state. The typical pattern reveals low serine, glycine, and 5-methyltetrahydrofolate levels in the cerebrospinal fluid. Another essential feature of this encephalopathy is that plasma serine and glycine levels are normal. Treatment with L-serine leads to a reduction in seizure burden [62,65].
Patients present with tonic spasms: Otahara syndrome: The primary seizure type in OS are epileptic spasms, observed in neonates with cortical dysgenesis. Usually, it has a later onset compared to EIMEE, but it can also start in the neonatal period.
Infants acutely develop tonic spasms that can be diffuse or lateralized, occur singly or in clusters, and are independent of sleep cycle. Spasms typically last up to 5 s and can occur hundreds of times daily [62,65]. Approximately one-third of patients with OS also develop other seizure types such as focal motor seizures, hemiconvulsions, or generalized tonic-clonic seizures [22,49]. The EEGs in OS indicate a suppression burst pattern comprising bursts of high-amplitude spikes and polyspike that alternates regularly with periods of electric suppression (Figure 3A and 3B). These bursts coincided with tonic spasms (Figures 3B and 3C). Typically, the burst suppression pattern remains unchanged during both wakefulness and sleep [22,49].
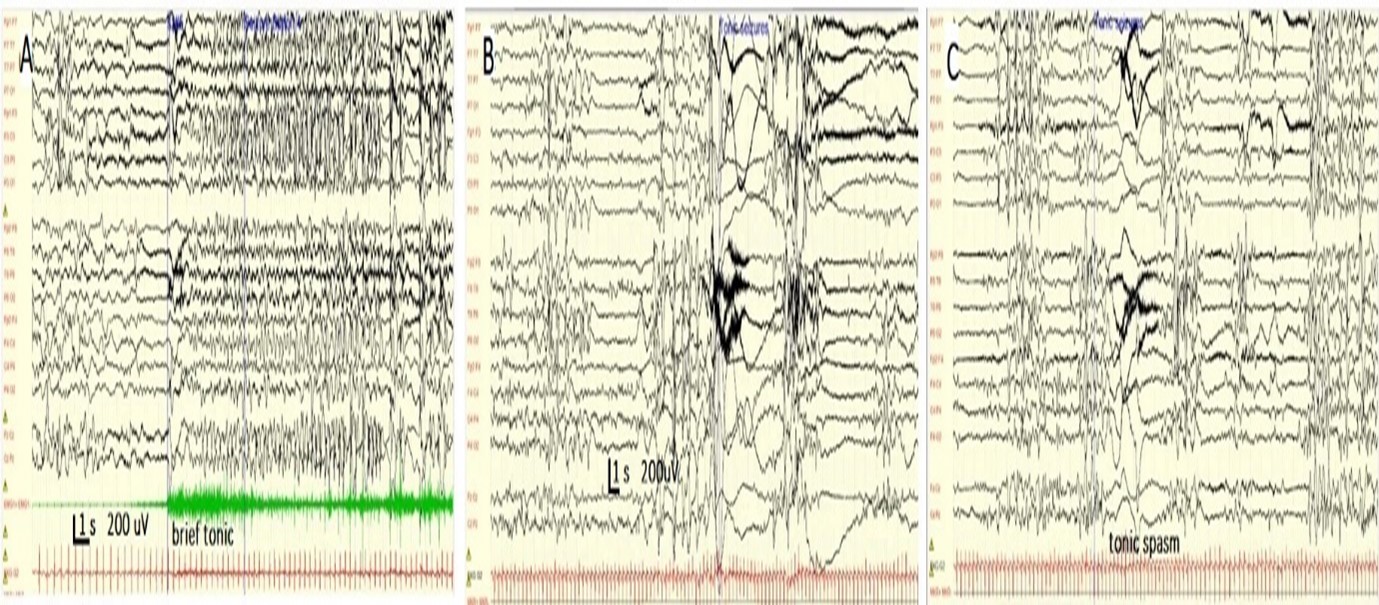
Figure 3. Different tonic seizures in neonates. A and B patients with Otahara syndrome. See the BS pattern and generalized suppression with superimposed fast activity during ictal tonic spasms. Patient C with SCNA8-DEE showed a sequential seizure, tachycardia [heart frequency increased from 167 to 221 LPM, followed by a tonic seizure).
Treatment
Neonatal seizure treatment follows a standard protocol in most institutions. This protocol is not magic but usually includes Phenobarbital IV with a loading dose (LD) of 20 mg/kg and maintenance dose (MD) of 2.5-3 mg/kg/day, Fosphenytoin/phenytoin IV LD of 20 mg/kg and MD of 2.5-3 mg/kg, Midazolam IV bolus 0.05 mg/kg twice a day or IV 0.05 mg/kg and if it is effective, then start infusion of 0.05 mg/kg/hour up to 0.5 mg/kg/hour [3,66,67]. Levetiracetam IV can be used at an LD of 40-60 mg/kg and MD of 30 mg/kg BID or Lidocaine IV LD of 2 mg/kg and 7 mg/kg/day every four h, which is reduced then to 3.5 mg/kg/hour during the following 12 h and then decreased to 0.75 mg/kg/hour per other 12 hours and stopped [68,69]. A recent randomized controlled trial documented that phenobarbital is superior to levetiracetam in the treatment of neonatal seizures [70]. Table 1 shows the personalized treatment of neonatal epilepsy based on the semiology and EEG results.
Conclusion
Strict electroclinical semiology is the backbone of neonatal seizure diagnosis. Acute symptomatic seizures should be distinguished from epileptic seizures. EEG and ictal semiology can help in diagnosis and provide a guide for genetic counseling and treatment. Neonatal seizure classification should be expanded to include the EEG patterns. With whole-exome sequence analysis, more genetic diseases causing epileptic encephalopathy are reported every day. Lumping them into a better classification system will prevent unnecessary and hazardous medication use.
Conflicts of Interest
The authors declare no conflicts of interest. The authors have no funding sources available for this study.
References
2. Pisani F, Spagnoli C. EEG in neonatal seizures: where to look and what to see. Expert Review of Neurotherapeutics. 2022;22(11-12):963-79.
3. Samanta D. Recent Advances in the Diagnosis and Treatment of Neonatal Seizures. Neuropediatrics. 2021 Apr;52(2):73-83.
4. Cilio MR, Sands TT. Neonatal-Onset Epilepsies: Early Diagnosis and Targeted Treatment. In: Perlman JM, Editor. Neurology: Neonatology Questions and Controversies. Netherlands: Elsevier Health Sciences; 2018.
5. Santarone ME, Pietrafusa N, Fusco L. Neonatal seizures: When semiology points to etiology. Seizure. 2020;80:161-5.
6. Biagioni E, Ferrari F, Boldrini A, Roversi MF, Cioni G. Electroclinical correlation in neonatal seizures. European Journal of Paediatric Neurology. 1998;2(3):117-25.
7. El Kosseifi C, Cornet MC, Cilio MR. Neonatal Developmental and Epileptic Encephalopathies. Semin Pediatr Neurol. 2019;32.
8. Pressler RM, Cilio MR, Mizrahi EM, Moshé SL, Nunes ML, Plouin P, et al. The ILAE Classification of Seizures the Epilepsies: Modification for Seizures in the Neonate. Proposal from the ILAE Task Force on Neonatal Seizures. Epilepsia. 2018;
9. Andrade E, Shaikh Z, Chavez W, Torres A. Treatment of neonatal seizures. Medicina (B Aires). 2018;78 Suppl 2:30-35.
10. Nagarajan L, Palumbo L, Ghosh S. Classification of clinical semiology in epileptic seizures in neonates. European Journal of Paediatric Neurology. 2012;16(2):118-25.
11. Kim EH, Shin J, Lee BK. Neonatal seizures: diagnostic updates based on new definition and classification. Clin Exp Pediatr. 2022;65(8):387-97.
12. Bariola MC. Neonatal seizures in therapeutic hypothermia era. Journal of Maternal-Fetal and Neonatal Medicine. 2021;34(SUPPL 1).
13. Costa J da, Nunes ML, Fiori RM. Seizures in the neonatal period. J Pediatr (Rio J). 2001; 2001 Jul:77 Suppl 1:S115-22.
14. Pressler RM, Cilio MR, Mizrahi EM, Moshé SL, Nunes ML, Plouin P, et al. The ILAE classification of seizures and the epilepsies: Modification for seizures in the neonate. Position paper by the ILAE Task Force on Neonatal Seizures. Epilepsia. 2021;62(3):615-28.
15. Ogihara M, Kinoue K, Takamiya H, Nemoto S, Miyajima T, Hoshika A, et al. A case of early infantile epileptic encephalopathy (EIEE) with anatomical cerebral asymmetry and myoclonus. Brain Dev. 1993;15(2):133-9.
16. Lombroso CT. Early myoclonic encephalopathy, early infantile epileptic encephalopathy, and benign and severe infantile myoclonic epilepsies: A critical review and personal contributions. Journal of Clinical Neurophysiology. 1990;7(3):380-408.
17. Pavone P, Corsello G, Ruggieri M, Marino S, Marino S, Falsaperla R. Benign and severe early-life seizures: A round in the first year of life. Vol. 44, Italian Journal of Pediatrics. 2018;44(1):54.
18. Cornet MC, Morabito V, Lederer D, Glass HC, Ferrao Santos S, Numis AL, et al. Neonatal presentation of genetic epilepsies: Early differentiation from acute provoked seizures. Epilepsia. 2021;62(8):1907-20.
19. Bayat A, Bayat M, Rubboli G, Møller RS. Epilepsy syndromes in the first year of life and usefulness of genetic testing for precision therapy. Genes. 2021;12(7):1051.
20. Spagnoli C, Fusco C, Percesepe A, Leuzzi V, Pisani F. Genetic neonatal-onset epilepsies and developmental/epileptic encephalopathies with movement disorders: A systematic review. International Journal of Molecular Sciences. 2021;22(8):4202.
21. Debnath B, Chowdhury RN, Shaha NC, Hussain ME. Epileptic Encephalopathies in Infants and Children: Study of Clinico-Electroencephalographic Spectrum in a Tertiary Hospital in Bangladesh. Open J Pediatr. 2021;11(03):339-50.
22. Fusco L, Pachatz C, Di Capua M, Vigevano F. Video/EEG aspects of early-infantile epileptic encephalopathy with suppression-bursts (Ohtahara syndrome). Brain and Development. 2001;23(7):708-14.
23. Ohtahara S. Seizure disorders in infancy and childhood. Brain Dev. 1984;6(6):509-19.
24. Mulkey SB, Ben-Zeev B, Nicolai J, Carroll JL, Grønborg S, Jiang YH, et al. Neonatal nonepileptic myoclonus is a prominent clinical feature of KCNQ2 gain-of-function variants R201C and R201H. Epilepsia. 2017;58(3):436-45.
25. Sands TT, Balestri M, Bellini G, Mulkey SB, Danhaive O, Bakken EH, et al. Rapid and safe response to low-dose carbamazepine in neonatal epilepsy. Epilepsia. 2016;57(12):2019-30.
26. Zhang Q, Li J, Zhao Y, Bao X, Wei L, Wang J. Gene mutation analysis of 175 Chinese patients with early-onset epileptic encephalopathy. Clin Genet. 2017;91(5):717-24.
27. Olson HE, Kelly M, LaCoursiere CM, Pinsky R, Tambunan D, Shain C, et al. Genetics and genotype–phenotype correlations in early onset epileptic encephalopathy with burst suppression. Ann Neurol. 2017;81(3):419-29.
28. Numis AL, Angriman M, Sullivan JE, Lewis AJ, Striano P, Nabbout R, et al. KCNQ2 encephalopathy: Delineation of the electroclinical phenotype and treatment response. Neurology. 2014;82(4):368-70.
29. Gürsoy S, Erçal D. Diagnostic Approach to Genetic Causes of Early-Onset Epileptic Encephalopathy. Journal of Child Neurology. 2016;31(4):523-32.
30. Harding BN. Progressive neuronal degeneration of childhood with liver disease (Alpers-Huttenlocher Syndrome): A personal review. Journal of Child Neurology. 1990;5(4):273-87.
31. Kelly M, Park M, Mihalek I, Rochtus A, Gramm M, Pérez-Palma E, et al. Spectrum of neurodevelopmental disease associated with the GNAO1 guanosine triphosphate–binding region. Epilepsia. 2019;60(3):406-18.
32. Bruun TUJ, Desroches CL, Wilson D, Chau V, Nakagawa T, Yamasaki M, et al. Prospective cohort study for identification of underlying genetic causes in neonatal encephalopathy using whole-exome sequencing. Genetics in Medicine. 2018;20(5):486-94.
33. Vilan A, Mendes Ribeiro J, Striano P, Weckhuysen S, Weeke LC, Brilstra E, et al. A Distinctive Ictal Amplitude-Integrated Electroencephalography Pattern in Newborns with Neonatal Epilepsy Associated with KCNQ2 Mutations. Neonatology. 2017;112(4):387-93.
34. Pisano T, Numis AL, Heavin SB, Weckhuysen S, Angriman M, Suls A, et al. Early and effective treatment of KCNQ2 encephalopathy. Epilepsia. 2015;56(5):685-91.
35. Millichap JJ, Park KL, Tsuchida T, Ben-Zeev B, Carmant L, Flamini R, et al. KCNQ2 encephalopathy: Features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol Genet. 2016;2(5):e96.
36. Fang Z xu, Zhang M, Xie L ling, Jiang L, Hong S qi, Li X juan, et al. KCNQ2 related early-onset epileptic encephalopathies in Chinese children. J Neurol. 2019;266(9):2224-32.
37. Vatta M, Tennison MB, Aylsworth AS, Turcott CM, Guerra MP, Eng CM, et al. A novel STXBP1 mutation causes focal seizures with neonatal onset. J Child Neurol. 2012;27(6):811-4.
38. Xian J, Parthasarathy S, Ruggiero SM, Balagura G, Fitch E, Helbig K, et al. Assessing the landscape of STXBP1-related disorders in 534 individuals. Brain. 2022;145(5):1668-83.
39. Swanson DA, Steel JM, Valle D. Identification and characterization of the human ortholog of rat STXBP1, a protein implicated in vesicle trafficking and neurotransmitter release. Genomics. 1998;48(3):373-6.
40. Wang QH, Cao JJ, Wang YY, Zhang MN, Liu LY, Wang J, et al. Efficacy of levetiracetam in STXBP1 encephalopathy with different phenotypic and genetic spectra. Seizure. 2022;95:64-74.
41. Fehr S, Wong K, Chin R, Williams S, De Klerk N, Forbes D, et al. Seizure variables and their relationship to genotype and functional abilities in the CDKL5 disorder. Neurology. 2016;87(21):2206-13.
42. Melikishvili G, Dulac O, Gataullina S. Neonatal SCN2A encephalopathy: A peculiar recognizable electroclinical sequence. Epilepsy and Behavior. 2020;111:107187.
43. Wolff M, Johannesen KM, Hedrich UBS, Masnada S, Rubboli G, Gardella E, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. 2017;140(5):1316-36.
44. Lu J, Zhao G, Lv D, Cao L, Zhao G. Autosomal dominant sleep-related hypermotor epilepsy associated with a novel mutation of KCNT1. Transl Neurosci. 2022;13(1):240-5.
45. Fehr C, Klein KM. Genetic syndromes associated with frontal lobe epilepsy. Zeitschrift fur Epileptologie. 2022;35:32-35.
46. McTague A, Appleton R, Avula S, Cross JH, King MD, Jacques TS, et al. Migrating partial seizures of infancy: Expansion of the electroclinical, radiological and pathological disease spectrum. Brain. 2013;136(5):1578-91.
47. Carapancea E, Cornet MC, Milh M, De Cosmo L, Huang EJ, Granata T, et al. Clinical and Neurophysiologic Phenotypes in Neonates with BRAT1 Encephalopathy. Neurology. 2023;100(12):e1234-47.
48. Piro E, Nardello R, Gennaro E, Fontana A, Taglialatela M, Mangano GD, et al. A novel mutation in KCNQ3-related benign familial neonatal epilepsy: electroclinical features and neurodevelopmental outcome. Epileptic Disorders. 2019;21(1):87-91.
49. Ohtahara S, Ohtsuka Y, Yamatogi Y, Oka E. The early-infantile epileptic encephalopathy with suppression-burst: Developmental aspects. Brain Dev. 1987;9(4):371-6.
50. Lucaccioni L, Righi B, Cingolani GM, Lugli L, Della Casa E, Torcetta F, et al. Overwhelming sepsis in a neonate affected by Zellweger syndrome due to a compound heterozygosis in PEX 6 gene: a case report. BMC Med Genet. 2020;21(1):229.
51. Celik M, Ipek MS, Ozgun N, Akdeniz O, Tuzun H, Bulbul A. Clinical diagnosis, biochemical findings, genetics and incidence of Zellweger syndrome. Iran J Pediatr. 2018;28(1):e58924.
52. Spiegel R, Schwahn BC, Squires L, Confer N. Molybdenum cofactor deficiency: A natural history. J Inherit Metab Dis. 2022;45(3):456-69.
53. Atwal PS, Scaglia F. Molybdenum cofactor deficiency. Molecular Genetics and Metabolism. 2016;117(1):1-4.
54. Tabatabaie L, Klomp LWJ, Rubio-Gozalbo ME, Spaapen LJM, Haagen AAM, Dorland L, et al. Expanding the clinical spectrum of 3-phosphoglycerate dehydrogenase deficiency. J Inherit Metab Dis. 2011;34(1):181-4.
55. Stockler S, Plecko B, Gospe SM, Coulter-Mackie M, Connolly M, van Karnebeek C, et al. Pyridoxine dependent epilepsy and antiquitin deficiency. Clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up. Molecular Genetics and Metabolism. 2011;104(1-2):48-60.
56. Kaminiów K, Pająk M, Pająk R, Paprocka J. Pyridoxine-Dependent Epilepsy and Antiquitin Deficiency Resulting in Neonatal-Onset Refractory Seizures. Brain Sciences. 2022;12(1):65.
57. Hansen J, Snow C, Tuttle E, Ghoneim DH, Yang CS, Spencer A, et al. De Novo Mutations in SIK1 Cause a Spectrum of Developmental Epilepsies. The American Journal of Human Genetics. 2015;96(4):682-90.
58. Mignon-Ravix C, Milh M, Kaiser CS, Daniel J, Riccardi F, Cacciagli P, et al. Abnormal function of the UBA5 protein in a case of early developmental and epileptic encephalopathy with suppression-burst. Hum Mutat. 2018;39(7):934-8.
59. Kim YO, Yang JH, Park C, Kim SK, Kim MK, Shin MG, et al. A novel PIGA mutation in a family with X-linked, early-onset epileptic encephalopathy. Brain Dev. 2016;38(8):750-4.
60. Lin W De, Chou IC, Tsai FJ, Hong SY. A novel PIGA mutation in a Taiwanese family with early-onset epileptic encephalopathy. Seizure. 2018;58:52-4.
61. André M V., Cacciagli P, Cano A, Vaugier L, Roussel M, Girard N, et al. The phenotype caused by recessive variations in SLC25A22: Report of a new case and literature review. Archives de Pediatrie. 2021;28(1):87-92.
62. Zara F, Gennaro E, Vari S, Coviello D, Striano P. OP12 – 2577: Targeted resequencing in epileptic encephalopathies: diagnostic implications and genotype–phenotype correlations. European Journal of Paediatric Neurology. 2015;19(Suppl 1):S4-S5.
63. Santalucia R, Vilain C, Soblet J, De Laet C, Vuckovic A, König J, et al. Carbamazepine efficacy in a severe electro-clinical presentation of SLC13A5-epilepsy. Ann Clin Transl Neurol. 2022;9(7):1095-99.
64. Matricardi S, De Liso P, Freri E, Costa P, Castellotti B, Magri S, et al. Neonatal developmental and epileptic encephalopathy due to autosomal recessive variants in SLC13A5 gene. Epilepsia. 2020;61(11):2474-85.
65. Rapaccini V, Miconi F, Esposito S, Pasini A. Pathogenetic Potential of the Mutations of SPTAN 1. J Hum Clin Genet. 2019;1(1):1-2.
66. Stevenson NJ, Vanhatalo S. Designing a trial for neonatal seizure treatment. Seminars in Fetal and Neonatal Medicine. 2018;23(3):213-7.
67. Pavel AM, Rennie JM, de Vries LS, Blennow M, Foran A, Shah DK, et al. Neonatal Seizure Management: Is the Timing of Treatment Critical? Journal of Pediatrics. 2022;243:61-8.e2.
68. Ahrens S, Ream MA, Slaughter LA. Status Epilepticus in the Neonate: Updates in Treatment Strategies. Current Treatment Options in Neurology. 2019;21(2):8.
69. Yamamoto H, Aihara M, Niijima S, Yamanouchi H. Treatments with midazolam and lidocaine for status epilepticus in neonates. Brain Dev. 2007;29(9):559-64.
70. Qiao MY, Cui HT, Zhao LZ, Miao JK, Chen QX. Efficacy and Safety of Levetiracetam vs. Phenobarbital for Neonatal Seizures: A Systematic Review and Meta-Analysis. Frontiers in Neurology. 2021;12:747745.
71. Kuersten M, Tacke M, Gerstl L, Hoelz H, Stülpnagel CV, Borggraefe I. Antiepileptic therapy approaches in KCNQ2 related epilepsy: A systematic review. European Journal of Medical Genetics. 2020;63(1):103628.
72. Kim HJ, Yang D, Kim SH, Kim B, Kim HD, Lee JS, et al. The phenotype and treatment of SCN2A-related developmental and epileptic encephalopathy. Epileptic Disorders. 2020;22(5):563-70.
73. Brunklaus A, Brünger T, Feng T, Fons C, Lehikoinen A, Panagiotakaki E, et al. The gain of function SCN1A disorder spectrum: Novel epilepsy phenotypes and therapeutic implications. Brain. 2022;145(11):3816-31.
74. Fitzgerald MP, Fiannacca M, Smith DM, Gertler TS, Gunning B, Syrbe S, et al. Treatment Responsiveness in KCNT1-Related Epilepsy. Neurotherapeutics. 2019;16(3):848-57.
75. Barker BS, Ottolini M, Wagnon JL, Hollander RM, Meisler MH, Patel MK. The SCN8A encephalopathy mutation p.Ile1327Val displays elevated sensitivity to the anticonvulsant phenytoin. Epilepsia. 2016;57(9):1458-66.
76. Boerma RS, Braun KP, van de Broek MPH, van Berkestijn FMC, Swinkels ME, Hagebeuk EO, et al. Remarkable Phenytoin Sensitivity in 4 Children with SCN8A-related Epilepsy: A Molecular Neuropharmacological Approach. Neurotherapeutics. 2016;13(1):238.
77. Dilena R, Striano P, Traverso M, Viri M, Cristofori G, Tadini L, et al. Dramatic effect of levetiracetam in early-onset epileptic encephalopathy due to STXBP1 mutation. Brain Dev. 2016;38(1):128-31.
78. Kessi M, Chen B, Shan LD, Wang Y, Yang L, Yin F, et al. Genotype-phenotype correlations of STXBP1 pathogenic variants and the treatment choices for STXBP1-related disorders in China. BMC Med Genomics. 2023;16(1):46.
79. Knight EMP, Amin S, Bahi-Buisson N, Benke TA, Cross JH, Demarest ST, et al. Safety and efficacy of ganaxolone in patients with CDKL5 deficiency disorder: results from the double-blind phase of a randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2022;21(5):417-27.
80. Giacomini T, Pisciotta L, Prato G, Meola I, Zara F, Fiorillo C, et al. Severe early-onset developmental and epileptic encephalopathy (DEE) associated with novel compound heterozygous pathogenic variants in SLC25A22: Case report and literature review. Seizure. 2019;70:56-8.
81. Coughlin CR, Tseng LA, Abdenur JE, Ashmore C, Boemer F, Bok LA, et al. Consensus guidelines for the diagnosis and management of pyridoxine-dependent epilepsy due to α-aminoadipic semialdehyde dehydrogenase deficiency. J Inherit Metab Dis. 2021;44(1):178-92.