Abstract
Alzheimer’s disease (AD) represents the most common cause of dementia. Even if AD is commonly viewed as a disorder primarily of memory, there are several other additional domains, including visual function. Accumulating evidence suggests that the magnocellular or M-pathway subsystem of visual pathways is preferentially affected in AD compared to other neurodegenerative disease. A study of Sartucci et al. few years ago tested the three main visual pathways subsystem electrophysiologically in AD patients. They used Pattern Electroretinograms (PERG) and Visual Evoked Potentials (VEP) to luminance-contrast and chromaticcontrast stimuli to elicit responses differentially driven by generators of M-pathway, P-pathway (Parvovellular) and K-pathway (Koniocellular), and found that responses driven by the M-pathway were relatively more altered. The results of Sartucci et al., together with the body of available literature, are consistent with the hypothesis that a primary dysfunction of M-pathway impairment does occur in a sufficient number of AD patients to warrant support for a specific Magnocellular deficit associated with this disease.
Keywords
Magnocellular stream, Alzheimer’s disease, Visual pathways subsystem, Chromatic and luminance visual evoked responses
Introduction
Alzheimer’s Disease (AD) is a progressive chronic neurodegenerative disorder and the most common cause of dementia in the older adults worldwide [1]. AD is commonly viewed as a disorder primarily of memory, which represents usually the initial sign; moreover, this disease is characterized by impairment in several additional domains, including visual function. However, the NINCDS-ADRDA clinical diagnostic criteria [2] make no mention of sensory changes. Several main fields of researches on vision in AD currently have undertaken, from structure to function and behavior at this time, and current directions in AD vision studies are extensively reported in many papers, including some exhaustive books [1].
This commentary will briefly review current knowledge on dysfunction of the visual pathway in AD, focusing on differential involvement of M, P, and K visual subsystems at either retinal or cortical levels. Visual dysfunction in AD will be compared visual dysfunction occurring in physiological aging and other age-related degenerative disorders.
Clinical Aspects
Alzheimer’s disease (AD) impairs visual function at an early-intermediate state of degeneration and functional losses correlate with severity of cognitive progressive impairment. Different visual deficits have been reported in AD patients, such as alteration of contrast sensitivity [3], motion perception [4,5], performance on masking tests [3], color discrimination of blue, short-wavelength hues [3], visual attention and visual behavior, face discrimination, imagery [6]. However further sensory and motor dysfunction in AD patients may be detected [7]. In the absence of an accurate biomarker, the diagnosis relies on cognitive profile supported by neuroimaging [8]. Following the abovementioned reports, the eye as has been proposed as a convenient biomarker for diagnosis and progression of AD [9,10]. Visual involvement features in AD, together with functional and morphological changes of the retina and visual structures, have received a great deal of attention [11].
Neuropathological Changes on Alzheimer’s Disease: Focus on the Visual System
Extensive deposition of amyloid-β (Aβ) leading to the formation of plaques is considered one of the key pathological features of Alzheimer’s disease (AD) [16]. However, impairment of memory and synaptic deficit occur prior to extensive extracellular deposition of Aβ in the brain [17-21] during an early phase of AD progression before senile plaques (SP) [22]. Interestingly, Origlia et al. [23] demonstrated that low concentrations of oligomeric Aβ are capable of disrupting synaptic plasticity in different circuits of the mouse visual cortex. An intriguing finding is that long-term synaptic plasticity (LTP) in supragranular horizontal pathways (layer II/ III) is altered by lower concentration of Aβ respect to other intracortical pathways (WM-layer II/III vertical pathway). Thus, sensitivity of synaptic disruption to oligomeric Aβ accumulation differs between different intra-cortical pathways within the occipital cortex.
The reported results, together with previous reports [24,25] showing the presence of laminar differences within single brain areas in AD progress, raise the important question of whether the pattern of AD reflects the sensitivity of different neurons and/or synaptic connections to pathogens, including Aβ.
Alterations in amyloid precursor protein (APP) processing and Aβ accumulation may therefore contribute to retinal damage as well as to cortical alterations as above indicated. Several reports outlined the role of oligomeric Aβ in retinal pigmented epithelium (RPE) alterations and dysfunctions, ganglion cells dysfunction, inflammatory and oxidative process till cell death [26,27]; indeed, APP and its products including Aβ were detected in retinal as well as in cerebral extracts from murine models of AD.
Altogether, the reported results permit to advance the hypothesis that visual system, from retina to cortical areas, might be affected during an early-intermediate stage of AD characterized by large absence of SP, NFT and cell death; following this idea an increasing amount of oligomeric beta accumulation induces synaptic toxicity and dysfunction in several cells from retina to cortex. Whether Aβ-dependent visual impairment in AD is due to retinal pathology or due to post-retinal pathology remains unclear. Therefore, the eye may offer a direct window to the cerebral pathology sharing many similarities to the brain [9].
Neuropathological Changes in Alzheimer’s Disease: The Visual Variant
In addition to the typical form of AD, a growing number of cases of focal cortical presentations has recently been described: in particular, the condition known as posterior cortical atrophy (PCA) is commonly accompanied by visuospatial impairment, which recalls the clinical characteristics of both Balint’s and Gerstmann’s Syndromes, visual agnosia, dressing apraxia, environmental disorientation. PCA presents as bi-parietal syndrome and tends to remain confined to posterior areas for a very long time before spreading away [12,13]. Patients with PCA are older than those with typical AD and have less memory loss or reduced verbal fluency. Unfortunately, differential diagnosis remains anamnestic, even if modern functional imaging, such as amyloid binding ligand PIB or positron emission tomography with carbon 11 [14], could be helpful to discriminate these pathologies in vivo. Moreover, the distribution of senile plaques (SP) and neurofibrillary tangles (NFT) is quite different between typical AD and visuospatial variant. Areas 17, 18 and 19 contain a much higher density of NFT in PCA; in patients with visual agnosia also the regions of inferior temporal cortex and occipito-temporal junction (areas 37, 20 and 21; [15]) show increased deposition of NFT and SP compared to typical AD. These data are consistent with the view that PCA affects the feed-forward projections to highlevel regions and to associative visual areas, but the etiopathological bases remain elusive so far.
Segregation of P- and M-stream in Humans: The Ventral (P) and Dorsal (M) Cortical Stream
The post-receptoral visual pathways of primates contain two major parallel streams specific for color contrast/ form discrimination and luminance contrast/movement discrimination [28-31]. The color-opponent system originates from small, tonic ganglion cells relaying to parvocellular laminae of the lateral geniculate nucleus and then projecting to layer 4C-β of the striate cortex. More deeply, recent data showed that two color-opponent pathways, red-green (RG) and blue-yellow (B-Y), form the so-called parvo- (P) and konio-cellular (K) streams, respectively [32-35]. The second major subsystem, i.e. the achromatic stream, originates from large, phasic ganglion cells projecting to the magnocellular (M) layers of the lateral geniculate nucleus and then to laminae 4C-β of the striate visual cortex [36,37]. It is worth to note that parvocellular cells may also respond well to achromatic contrast stimuli of relatively high-spatial frequency. However, within the range of spatial frequencies to which both streams respond, M cells are relatively more sensitive to achromatic contrast and this characteristic is more prominent at higher temporal frequencies. To date, it is common knowledge that a specific decline in highspatial contrast and low-temporal frequency sensitivity suggests a selective deficit in P-stream [38], whereas the loss of motion perception and the impairment in analyzing low-spatial frequency and contrast luminance are more likely to be related with severe dysfunction affecting M-pathway. The small receptive fields of the P channels in the center of vision yield a strong local motion signal, whereas the M ones with their fast transmission time and large receptive fields ensure the integration of global motion signals over a larger area. At higher level P inputs mainly project to the ventral pathway, whereas K and M pathways to the dorsal pathway [39].
Since parietal structures are a frequent pathophysiological target in AD, a dorsal stream visual impairment (M and K pathways) might be predominantly affected and occurs independently in AD [40].
From Histopathology to Psychophysiology: New Insights into the Involvement of Magnocellular Stream in Alzheimer’s Disease
Histopathological studies demonstrated that hippocampus and the parahippocampal regions are the earliest affected regions in AD, suggesting a hierarchical order of involvement: from limbic to associative cortical areas, from associative to primary sensory cortical areas including the visual cortex. Following this scheme, the involvement of visual system characterizes a late phase of AD, as well as the non-pathological aging [41]. However, recent studies have highlighted visual dysfunctions in AD patients. In particular, clinical studies support a link between cognitive performance and visual dysfunction even at an early stage of AD when SP and NFT are largely absent. Impairment of higher order visual processing, such as the loss of color discrimination, stereoacuity, contrast sensitivity, and backward masking, have been frequently described in AD patients [3,42,43].
Blanks and colleagues first tried to examine the degeneration of the retinal ganglion cells, finding that cells with largest diameters, by which magnocellular stream is made up, are selectively affected in AD patients [44]. Another indirect, but highly convincing data in support of a specific visual impairment come from the study of Dentchev et al. [45] who emphasized the presence of Aβ proteins in the retina of AD patients compared to age and sex-matched controls.
Hof and Morrison hypothesized that, even if there is less NFT deposition in the occipital areas than in the prefrontal and temporal cortices, AD patients often exhibit a significant neuronal loss within specific cortical layers, i.e. IV-C and IV-B, where magnocellular stream terminates [46]; this observation agrees with both a motion perception impairment and an extensive M-pathway dysfunction in AD, possibly caused by oligomeric Aβ progressive accumulation and sensitivity in different cortical layers [23].
Author Experience
The paper of Sartucci et al. [47] tested the hypothesis that AD involves a deficit in the magnocellular pathway of the visual system using ERGs and VEPs to chromatic (Ch, P and K streams) and luminance (Lum) stimuli that differentially drive responses from different subsystems. Electrophysiological analysis of these three different streams might be relatively simple also considering that sensory inputs are conveyed to the LGN, and then to V1 cortical area and higher center, in a one-to-one ordered fashion.
To this aim, they recorded electroretinograms and visual potentials evoked by chromatic and luminance visual stimuli (ChPERGs and ChVEPs, LumERGs and LumVEPs) in a selected sample of AD patients. All cases met the diagnostic criteria of probable AD according to the Diagnostic and Statistical Manual of Mental Disorders, fourth edition (DSM-4), the ICD-10 Classification of Mental and Behavioral Disorders (ICD-10) and the criteria of the National Institute of Neurologic and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDSADRDA) [48]. Severity of dementia was assessed using MMSE [49], and an extensive standardized diagnostic protocol [50]; at the time of diagnosis, all the patients presented a MMSE score below 23/30. Diagnosis of AD was supported by an exhaustive diagnostic work-up including morphological and functional neuroimaging (brain MRI scan and a FDG-PET semi quantitative study, electroencephalogram and auditory P300 Event Related Potentials (ERPs).
Visual stimuli
Visual stimuli were equiluminant horizontal sinusoidal gratings, modulated either in luminance (yellow-black, Y-Bk) or chromaticity (red-green, R-G and blue-yellow, B-Y). Details of stimuli, modality of recordings and device used are widely illustrated and reported in several papers (for example in Porciatti & Sartucci [34,35], Sartucci & Porciatti [51], Bonfiglio et al. [52]).
Results
Individual transient waveforms and grand-average of PERGs and VEPs, obtained in both controls and AD patients, are summarized in Figures 1 and 2. Compared to controls, Lum PERGs were inconsistent in individual patients, resulting in an altered grand-average waveform with reduced amplitude and delayed latency, suggesting the existence of a specific magnocellular or M-pathway deficit. Chromatic PERGs did not show obvious differences between controls and AD patients. To assess retinal versus post-retinal defects, Retino-cortical time (RCT) analysis was performed and did not reveal a retinocalcarine pathway involvement associated with AD [47].
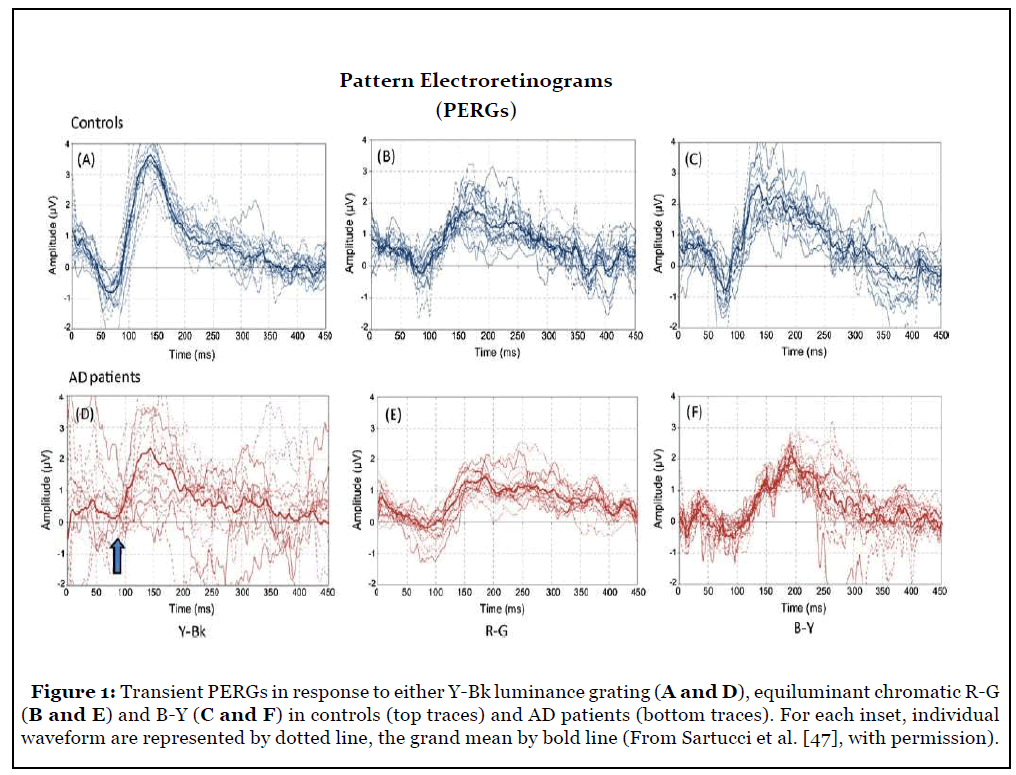
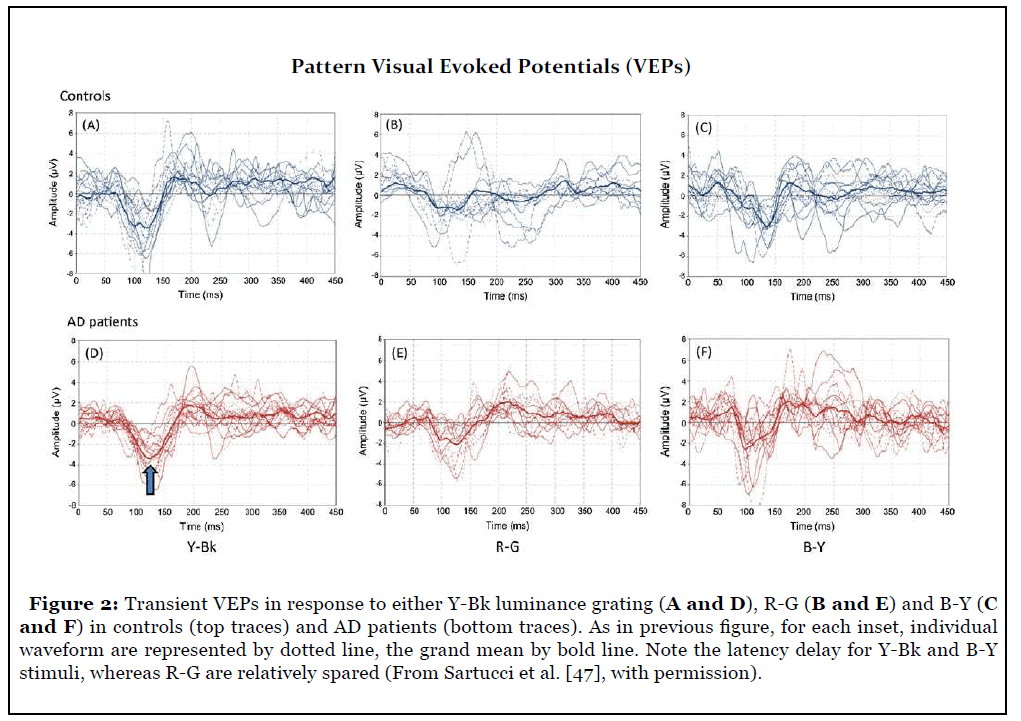
Locus of Visual Impairment As a Primary Marker of Alzheimer’s Disease
Evidence supporting a “magnocellular deficit hypothesis” was not previously considered. Rizzo et al. argued that visual impairment in AD is totally due to dysfunction in the associative cortices, basing their theory on normality of VEPs, full-field ERG and cortical flicker fusion threshold in AD [43,53]. On the other hand, the sensory deficits can be hidden and frequently masqueraded as higher order deficits.
Tippett et al. [54] investigated high-level visual processing involved in mild-to-moderate AD: they hypothesized two distinct pathways of visual processing altered in mild-to-moderate AD, basic shape perception and face processing. To rule out a primary, high-order visual impairment in AD it’s worth considering that (1) there is no relation between visual task performance and MMSE score and (2) truly segregated and symmetrical opponent-color systems do not exist in the cortex, where both luminance information and chromatic contrasts are simultaneously processed to detect spatial patterns [55]. Another interesting issue is whether the two hemispheres are asymmetrically involved in AD patients: in fact, the right hemisphere is dominant for rod-mediated scotopic behavior, whereas the left one may be dominant for conemediated photopic behavior [56].
Making the Difference between Central and Peripheral Visual Involvement in Alzheimer’s Disease
In the past, the reason for a prudent interpretation of previously reported evidences is that there are many contradictory signs of a specific visual involvement: shrinkage of both P and M ganglion cells in the retina, thinning of the nerve fiber layer [57], as well as pathology in both the magno- and the parvocellular layers of lateral geniculate nucleus (LGN), without any definitive conclusion on the selective vulnerability of a visual subsystem [58]. Some authors concluded that the unspecific alterations in second-order neurons pool reflects an initial age-dependent photoreceptors dysfunction [59]. Since age-dependent modifications were substantially the same for sensitivity to luminance and color contrast [60,61], it has been argued that they all arise at a peripheral level, with obvious reference to senile miosis, increased intraocular light scatter, decreased retinal blood flow in narrow veins or opacification of the ocular media [41], promoted by Aβ proteins aggregation. Some authors have also described a disease-related enlargement of the optic nerve head cupping as well as a marked thinning of neuroretinal rim [57]. For instance, it cannot be excluded that the outer retina may show some signs of impairment in AD patients: in fact, age-dependent morphological alterations in secondorder retinal neurons is commonly accompanied by dendrites loss, redistribution of glutamate receptors and simplification of horizontal cells network [62,63]. These all processes are characterized by a clear spatial gradient, increasing in number from the centre to the periphery of the retina. However, it’s worth remembering that compensatory remodeling, in relation to gradual increase in both ectopic synapses and collateral sprouting, follows a similar trend from the periphery, where magnocellular pathway arises, to foveal and parafoveal areas [63]. Besides, it is likely that retinal ganglion cell loss may be partially secondary to retrograde axonal degeneration too, since there’s a greater degeneration in the posterior optic nerve [64]. Taken together, these evidences play against a peripheral source of visual impairment in AD, consistent with the findings that AD particularly affects either cortico-cortical projection neurons [65,66] or geniculo-calcarine processing.
Conclusions
Results of Sartucci et al [47] in AD patients show evident abnormalities both in latency and amplitude of LumPERGs, and no significant changes in ChPERGs compared to controls, suggesting that the M-stream is the best candidate to explain impairments of visual function in this disease. In previous AD studies, it was hypothesized an aspecific deficit in primary visual processing and selective in secondary [67,68]; interestingly, Moore outlined the utility of using a black and a white monitor with large pattern and high contrast in establishing the diagnosis of AD [68], which appears to primarily drive response of the M-pathway. In agreement with Sartucci et al. [47], dysfunction of M-pathway in AD was also hypothesized and specified by Jacob et al. [69]. A specific early dorsal stream visual impairment independently has been reported using electrophysiological [70] or psychophysical [40] methods. Additional clinical and neuropathological evidence in AD supports involvement of the peripheral magnocellular stream or, in cortex, of the dorsal processing stream, supporting a formative theory that propose exceptional vulnerability of functions associated with the magnocellular pathway in AD.
It is worth noting that in another neurodegenerative pathology such as early idiopathic Parkinson’s Disease, both chromatic-contrast and luminance-contrast VEPs are impaired, with no evidence of specific involvement of visual subsystems [51].
Abnormal M pathway function has been reported also in subjects with autism spectrum disorders [71,72] a condition known to include cognitive and behavioral development impairments [73], even if later disputed [ 74].
Altogether, even if the results obtained in AD patients using electrophysiological responses are interesting and support a preferential involvement of the M-pathway, the theme remains debated and not definitively solved. Further studies with larger sample sizes will be necessary to confirm these results and expanded to include other type of degenerative diseases and additional forms of vision testing; it is also desirable that a better understanding of vision-related magnocellular early impairment could guide interventions to improve functional abilities in AD patients with benefit for either patients, their family, caregivers and the community.
Acknowledgment
We are grateful to Carlo Orsini for his technical help in implementing set of stimuli and assistance in recording patients with AD. Research in the Clinical Neurophysiopathology Unit laboratory was supported by the research funds of the Pisa University.
References
2. Tierney MC, Fisher RH, Lewis AJ, Zorzitto ML, Snow WG, Reid DW, et al. The NINCDS-ADRDA Work Group criteria for the clinical diagnosis of probable Alzheimer’s disease: a clinicopathologic study of 57 cases. Neurology. 1988 Mar 1;38(3):359-64.
3. Cronin-Golomb AL, Rizzo JF, Corkin S, Growdon JH. Visual Function in Alzheimer’s Disease and Normal Aging a. Annals of the New York Academy of Sciences. 1991 Dec;640(1):28-35.
4. Gilmore GC, Wenk HE, Naylor LA, Koss E. Motion perception and Alzheimer’s disease. Journal of Gerontology. 1994 Mar 1;49(2):P52-7.
5. Duffy CJ, Tetewsky SJ, O’Brien H. Cortical motion blindness in visuospatial AD. Neurobiology of Aging. 2000 Nov 1;21(6):867-9.
6. Valenti D. The anterior visual system and circadian function with reference to Alzheimer’s disease. InVision in Alzheimer’s Disease 2004 (Vol. 34, pp. 1-29). Karger Publishers.
7. Albers MW, Gilmore GC, Kaye J, Murphy C, Wingfield A, Bennett DA, et al. At the interface of sensory and motor dysfunctions and Alzheimer’s disease. Alzheimer’s & Dementia. 2015 Jan 1;11(1):70-98.
8. Scheltens P, Kittner B. Preliminary results from an MRI/CT-based database for vascular dementia and Alzheimer’s disease. Annals of the New York Academy of Sciences. 2000 Apr;903(1):542-6.
9. Lim JK, Li QX, He Z, Vingrys AJ, Wong VH, Currier N, et al. The eye as a biomarker for Alzheimer’s disease. Frontiers in Neuroscience. 2016 Nov 17;10:536.
10. Criscuolo C, Fabiani C, Cerri E, Domenici L. Synaptic dysfunction in Alzheimer’s disease and glaucoma: from common degenerative mechanisms toward neuroprotection. Frontiers in Cellular Neuroscience. 2017 Feb 27;11:53.
11. Cerquera-Jaramillo MA, Nava-Mesa MO, González- Reyes RE, Tellez-Conti C, de-la-Torre A. Visual features in Alzheimer’s disease: from basic mechanisms to clinical overview. Neural Plasticity. 2018;2018.
12. Alladi S, Xuereb J, Bak T, Nestor P, Knibb J, Patterson K, et al. Focal cortical presentations of Alzheimer’s disease. Brain. 2007 Oct 1;130(10):2636-45.
13. Maia da Silva MN, Millington RS, Bridge H, James- Galton M, Plant GT. Visual dysfunction in posterior cortical atrophy. Frontiers in Neurology. 2017 Aug 16;8:389.
14. Ng SY, Villemagne VL, Masters CL, Rowe CC. Evaluating Atypical Dementia Syndromes Using Positron Emission Tomography With Carbon 11–Labeled Pittsburgh Compound B. Archives of Neurology. 2007 Aug 1;64(8):1140-4.
15. von Guntena A, Giannakopoulosa P, Bourasb C, Hofc PR. Neuropathological Changes in Visuospatial Systems in Alzheimer’s. Vision in Alzheimer’s disease. 2004;34:30.
16. Gandy S. The role of cerebral amyloid ß accumulation in common forms of Alzheimer disease. The Journal of Clinical Investigation. 2005 May 2;115(5):1121-9.
17. Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice”. Science 274: 99-102. Hsiao, K.(1998). Transgenic mice expressing Alzheimer amyloid precursor proteins”. Experimental Gerontology. 1996;33:883-9.
18. Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, et al. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proceedings of the National Academy of Sciences. 1999 Mar 16;96(6):3228-33.
19. Redwine JM, Kosofsky B, Jacobs RE, Games D, Reilly JF, Morrison JH, et al. Dentate gyrus volume is reduced before onset of plaque formation in PDAPP mice: a magnetic resonance microscopy and stereologic analysis. Proceedings of the National Academy of Sciences. 2003 Feb 4;100(3):1381-6.
20. Arancio O, Zhang HP, Chen X, Lin C, Trinchese F, Puzzo D, et al. RAGE potentiates Aß-induced perturbation of neuronal function in transgenic mice. The EMBO journal. 2004 Oct 13;23(20):4096-105.
21. Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, et al. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences. 2006 Mar 28;103(13):5161-6.
22. Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002 Oct 25;298(5594):789-91.
23. Origlia N, Capsoni S, Cattaneo A, Fang F, Arancio O, Yan SD, et al. Aß-dependent inhibition of LTP in different intracortical circuits of the visual cortex: the role of RAGE. Journal of Alzheimer’s Disease. 2009 Jan 1;17(1):59-68.
24. Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL. Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science. 1984 Sep 14;225(4667):1168-70.
25. Gómez-Isla T, Price JL, McKeel Jr DW, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. Journal of Neuroscience. 1996 Jul 15;16(14):4491- 500.
26. Bruban J, Glotin AL, Dinet V, Chalour N, Sennlaub F, Jonet L, et al. Amyloid-ß (1-42) alters structure and function of retinal pigmented epithelial cells. Aging cell. 2009 Apr;8(2):162-77.
27. Dutescu RM, Li QX, Crowston J, Masters CL, Baird PN, Culvenor JG. Amyloid precursor protein processing and retinal pathology in mouse models of Alzheimer’s disease. Graefe’s Archive for Clinical and Experimental Ophthalmology. 2009 Sep 1;247(9):1213-21.
28. De Monasterio FM, Gouras P. Functional properties of ganglion cells of the rhesus monkey retina. The Journal of Physiology. 1975 Sep 1;251(1):167-95.
29. Dreher B, Fukada Y, Rodieck RW. Identification, classification and anatomical segregation of cells with X-like and Y-like properties in the lateral geniculate nucleus of old-world primates. The Journal of Physiology. 1976 Jun 1;258(2):433-52.
30. Shapley R. Visual sensitivity and parallel retinocortical channels. Annual Review of Psychology. 1990 Feb;41(1):635-58.
31. Van Essen DC, Gallant JL. Neural mechanisms of form and motion processing in the primate visual system. Neuron. 1994 Jul 1;13(1):1-0.
32. Merigan WH, Maunsell JH. How parallel are the primate visual pathways?. Annual Review of Neuroscience. 1993 Mar;16(1):369-402.
33. Dacey DM, Lee BB. The’blue-on’opponent pathway in primate retina originates from a distinct bistratified ganglion cell type. Nature. 1994 Feb;367(6465):731-5.
34. Porciatti V, Sartucci F. Retinal and cortical evoked responses to chromatic contrast stimuli: specific losses in both eyes of patients with multiple sclerosis and unilateral optic neuritis. Brain. 1996 Jun 1;119(3):723-40.
35. Porciatti V, Sartucci F. Normative data for onset VEPs to red-green and blue-yellow chromatic contrast. Clinical Neurophysiology. 1999 Apr 1;110(4):772-81.
36. Lennie P, Krauskopf J, Sclar G. Chromatic mechanisms in striate cortex of macaque. Journal of Neuroscience. 1990 Feb 1;10(2):649-69.
37. Engel S, Zhang X, Wandell B. Colour tuning in human visual cortex measured with functional magnetic resonance imaging. Nature. 1997 Jul;388(6637):68-71.
38. Di Russo F, Spinelli D. Spatial attention has different effects on the magno-and parvocellular pathways. Neuroreport. 1999 Sep 9;10(13):2755-62.
39. Collins E, Freud E, Kainerstorfer JM, Cao J, Behrmann M. Temporal dynamics of shape processing differentiate contributions of dorsal and ventral visual pathways. Journal of Cognitive Neuroscience. 2019 Jun;31(6):821- 36.
40. Lemos R, Figueiredo P, Santana I, Simões MR, Castelo-Branco M. Temporal integration of 3D coherent motion cues defining visual objects of unknown orientation is impaired in amnestic mild cognitive impairment and Alzheimer’s disease. Journal of Alzheimer’s Disease. 2012 Jan 1;28(4):885-96.
41. Fiorentini A, Porciatti V, Morrone MC, Burr DC. Visual ageing: unspecific decline of the responses to luminance and colour. Vision Research. 1996 Nov 1;36(21):3557-66.
42. Mendez MF, Tomsak RL, Remler B. Disorders of the visual system in Alzheimer’s disease. J Clin Neuroophthalmol. 1990 Mar 1;10(1):62-9.
43. Rizzo M, Anderson SW, Dawson J, Nawrot M. Vision and cognition in Alzheimer’s disease. Neuropsychologia. 2000 Jul 1;38(8):1157-69.
44. Blanks JC, Hinton DR, Sadun AA, Miller CA. Retinal ganglion cell degeneration in Alzheimer’s disease. Brain Research. 1989 Nov 6;501(2):364-72.
45. Dentchev T, Milam AH, Lee VM, Trojanowski JQ, Dunaief JL. Amyloid-ß is found in drusen from some agerelated macular degeneration retinas, but not in drusen from normal retinas. American Journal of Ophthalmology. 2003 May 14;136(4):787.
46. Hof PR, Bouras C, Constantinidis J, Morrison JH. Selective disconnection of specific visual association pathways in cases of Alzheimer’s disease presenting with Balint’s syndrome. Journal of neuropathology and Experimental Neurology. 1990 Mar 1;49(2):168-84.
47. Sartucci F, Borghetti D, Bocci T, Murri L, Orsini P, Porciatti V, Origlia N, Domenici L. Dysfunction of the magnocellular stream in Alzheimer’s disease evaluated by pattern electroretinograms and visual evoked potentials. Brain Research Bulletin. 2010 May 31;82(3-4):169-76.
48. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group* under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984 Jul 1;34(7):939-944.
49. Tognoni G, Ceravolo R, Nucciarone B, Bianchi F, Dell’Agnello G, Ghicopulos I, Siciliano G, Murri L. From mild cognitive impairment to dementia: a prevalence study in a district of Tuscany, Italy. Acta Neurologica Scandinavica. 2005 Aug;112(2):65-71.
50. Crum RM, Anthony JC, Bassett SS, Folstein MF. Population-based norms for the Mini-Mental State Examination by age and educational level. JAMA. 1993 May 12;269(18):2386-91.
51. Sartucci F, Porciatti V. VEPs to onset of chromatic red-green and blue-yellow gratings in Parkinson’s disesase never treated with L-Dopa. Journal of Clinical Neurophysiology. 2006 Oct;23(5):431-5.
52. Bonfiglio L, Bocci T, Minichilli F, Crecchi A, Barloscio D, Spina DM, et al. Defective chromatic and achromatic visual pathways in developmental dyslexia: Cues for an integrated intervention programme. Restorative Neurology and Neuroscience. 2017 Jan 1;35(1):11-24.
53. Rizzo JF, Cronin-Golomb A, Growdon JH, Corkin S, Rosen TJ, Sandberg MA, et al. Retinocalcarine function in Alzheimer’s disease: a clinical and electrophysiological study. Archives of Neurology. 1992 Jan 1;49(1):93-101.
54. Tippett LJ, Blackwood K, Farah MJ. Visual object and face processing in mild-to-moderate Alzheimer’s disease: from segmentation to imagination. Neuropsychologia. 2003 Jan 1;41(4):453-68.
55. Regan D. Evoked potentials and evoked magnetic fields in science and medicine. Human Brain Electrophysiology. 1989:59-61.
56. Braun CM, Achim A, Charron JF, Cote A. Dissociation of hemispheric exploitation of rods and cones for simple detection. American Journal of Psychology. 1998 Jul 1;111:241-64.
57. Berisha F, Feke GT, Trempe CL, McMeel JW, Schepens CL. Retinal abnormalities in early Alzheimer’s disease. Investigative Ophthalmology & Visual Science. 2007 May 1;48(5):2285-9.
58. Erskine D, Taylor JP, Firbank MJ, Patterson L, Onofrj M, O’Brien JT, et al. Changes to the lateral geniculate nucleus in A lzheimer’s disease but not dementia with L ewy bodies. Neuropathology and Applied Neurobiology. 2016 Jun;42(4):366-76.
59. Gresh J, Goletz PW, Crouch RK, Rohrer B. Structure– function analysis of rods and cones in juvenile, adult, and aged C57BL/6 and Balb/c mice. Visual Neuroscience. 2003 Mar;20(2):211-20.
60. Ruddock KH. The effect of age upon colour vision—II. changes with age in light transmission of the ocular media. Vision Research. 1965 Jan 1;5(1-3):47-58.
61. Suzuki TA, Qiang Y, Sakuragawa S, Tamura H, Okajima K. Age-related changes of reaction time and p300 for low-contrast color stimuli: effects of yellowing of the aging human lens. Journal of Physiological Anthropology. 2006;25(2):179-87.
62. Strettoi E, Porciatti V, Falsini B, Pignatelli V, Rossi C. Morphological and functional abnormalities in the inner retina of the rd/rd mouse. Journal of Neuroscience. 2002 Jul 1;22(13):5492-504.
63. Terzibasi E, Calamusa M, Novelli E, Domenici L, Strettoi E, Cellerino A. Age-dependent remodelling of retinal circuitry. Neurobiology of Aging. 2009 May 1;30(5):819-28.
64. Hinton DR, Sadun AA, Blanks JC, Miller CA. Opticnerve degeneration in Alzheimer’s disease. New England Journal of Medicine. 1986 Aug 21;315(8):485-7.
65. Hof PR, Morrison JH. Quantitative analysis of a vulnerable subset of pyramidal neurons in Alzheimer’s disease: II. Primary and secondary visual cortex. Journal of Comparative Neurology. 1990 Nov 1;301(1):55-64.
66. Kavcic V, Fernandez R, Logan D, Duffy CJ. Neurophysiological and perceptual correlates of navigational impairment in Alzheimer’s disease. Brain. 2006 Mar 1;129(3):736-46..
67. Brodie EE, Allan D, Brooks DN, McCulloch J, Foulds WS. Flash and pattern reversal visual evoked responses in normal and demented elderly. Cortex. 1992 Jun 1;28(2):289-93.
68. Moore NC. Visual evoked responses in Alzheimer’s disease: a review. Clinical Electroencephalography. 1997 Jul;28(3):137-42.
69. Jacob B, Hache JC, Pasquier F. Dysfunction of the magnocellular pathway in Alzheimer’s disease. Revue Neurologique. 2002 May;158(5 Pt 1):555-64.
70. Kubová Z, Kremlácek J, Vališ M, Langrová J, Szanyi J, Vít F, Kuba M. Visual evoked potentials to pattern, motion and cognitive stimuli in Alzheimer’s disease. Documenta ophthalmologica. 2010 Aug 1;121(1):37-49.
71. McCleery JP, Allman E, Carver LJ, Dobkins KR. Abnormal magnocellular pathway visual processing in infants at risk for autism. Biological Psychiatry. 2007 Nov 1;62(9):1007-14.
72. Koh HC, Milne E, Dobkins K. Contrast sensitivity for motion detection and direction discrimination in adolescents with autism spectrum disorders and their siblings. Neuropsychologia. 2010 Dec 1;48(14):4046-56.
73. LeBlanc JJ, Fagiolini M. Autism: a “critical period” disorder?. Neural plasticity. 2011;2011.
74. Fujita T, Yamasaki T, Kamio Y, Hirose S, Tobimatsu S. Parvocellular pathway impairment in autism spectrum disorder: Evidence from visual evoked potentials. Research in Autism Spectrum Disorders. 2011 Jan 1;5(1):277-85.