Abstract
Introduction: Cardiac amyloidosis is a chronic and progressive disease caused by the deposition of amyloid fibrils in cardiac tissues. The diagnosis and management of cardiac amyloidosis are complicated and have developed over the years.
Methods: An online survey was sent to cardiologists in four countries (Saudi Arabia, Lebanon, Egypt, and Iraq) interested in heart failure and had been practicing for more than a year. The survey included questions about the characteristics of the participants and their affiliated institutes. The survey addresses their knowledge and practices in cardiac amyloidosis concerning diagnostic modalities, treatment options, and interest in education and knowledge exchange.
Results: A total of 85 physicians participated in the online survey. Twenty-five were from Saudi Arabia, 24 from Lebanon, 24 from Egypt, and 12 from Iraq. There was a variation in the participating cardiologists’ knowledge, experience level, and readiness of their affiliated institutes to manage patients with ATTR-CM. Most participants believed that high rate of ATTR-CM misdiagnosis existed. Participants’ knowledge varied concerning the diagnostic modalities and ‘red flags’ raising suspicion about ATTR-CM. Another challenge was the availability of essential diagnostic modalities among various cardiology centers. A knowledge gap was also present regarding the updates in ATTR-CM management. However, there was a high endorsement towards the need for more education, physician networking, and knowledge exchange. Discussion and
Conclusion: This survey highlighted the need for increasing awareness levels among cardiologists in the four selected Arab countries. Cardiologists are most likely to benefit from additional training and knowledge exchange on the latest management advances of this disease. Thus, measures must be taken to focus on the physician’s awareness of ATTR-CM patient journey to achieve a better quality of care and outcome.
Keywords
Amyloidosis, Cardiac amyloidosis, Cardiology, Awareness, Arab countries, Diagnosis, Management, Cardiomyopathy
Introduction
Cardiac amyloidosis (CA) is a progressive disease affecting the normal cardiac structure and function [1]. CA could be isolated or associated with a spectrum of organ involvement, including the kidneys, lungs, nervous systems, bones, and others [2]. As the disease progresses, more amyloid fibrils deposit leading to increased stiffness, diastolic dysfunction, and ultimately congestive heart failure [3]. Cardiac involvement has a major impact on outcomes in patients with amyloidosis. The diagnosis of cardiac amyloidosis remains challenging, and early diagnosis is critical for patient management. The most common subtypes are light chain amyloidosis (AL) and transthyretin amyloidosis (ATTR) [4,5]. The exact prevalence rates of the disease are not well known, and the current numbers are thought to be a significant underreporting of the actual statistics rendering the disease undiagnosed for many years despite the recent advances in the non-invasive testing modalities to diagnose CA [6-8].
Cardiologists may lack specialization and expertise in diagnosing and managing ATTR, mainly due to the low frequency and long duration of diagnosis. Many patients require more than four years to reach a proper diagnosis [9,10]. In addition, misdiagnosis is very common with ATTR cardiomyopathy because of its similarity to many other cardiomyopathies and occurs in around 34 – 57% of patients [11]. All these factors are detrimental to the patients and lead to more advanced disease and worse disease outcomes [12].
Though minimal data have originated from Arab countries, it’s thought that the Arab world is no exception. Therefore, this study aims 1) to investigate the current awareness and knowledge of cardiac amyloidosis among cardiologists in four Arab countries, including Lebanon, Saudi Arabia, Egypt, and Iraq, and 2) to gather insights on the diagnostic modalities, treatment options, willingness for education and international collaborations of physicians managing patients with cardiomyopathies associated with amyloid transthyretin.
Methods
This is a cross-sectional non-interventional study conducted among 85 cardiologists in Lebanon, Saudi Arabia, Egypt, and Iraq. A 30-minute online survey was sent by email to all participants, including 36 questions. The survey included general (i) demographic questions, such as specialty, country, and affiliation, (ii) questions about knowledge of CA, (iii) practices in diagnosis and management of CA, (iv) their level of expertise in the field, and (v) their willingness and interests in having more education and training about CA.
Respondents were invited to participate using local opt-in panels of physicians. Physicians were sent an email invitation to participate in a research study on cardiac treatments. The study was self-administered using links provided by the survey’s web host. Screening questions at the beginning of the survey determined eligibility to participate. Only those who met all three inclusion criteria were advanced to the survey. All participating physicians were cardiologists affiliated with a local hospital or cardiac care facility and had knowledge of or a high level of interest in learning about cardiac amyloidosis.
Data was collected between October 26 and November 27, 2021. Survey participation was anonymous, voluntary, and in compliance with data protection laws in the aforementioned countries. Additional ethical approval from the corresponding Institutional Review Boards (IRB) was not required, as the study was a clinical practice survey that did not involve any patients or data identifiable to specific human subjects. Data was incorporated and analyzed on SPSS software version 25.0 (IBM Corporation Armonk, New York, USA). Categorical variables were reported using descriptive statistics (frequency and percentages), while continuous variables were reported as mean and standard deviation [SD]. The data is presented in tables and bar graphs.
Results
General characteristics of the survey participants
All 85 physicians completed the online survey. As reported in Table 1, fourteen were cardio-imaging physicians, 27 were heart failure cardiologists, and 44 were interventional cardiologists. Twenty-five physicians were from Saudi Arabia, 24 from Lebanon, 24 from Egypt, and 12 from Iraq. Around 71% of the physicians reported they attended several educational meetings, either in person or virtually, about amyloid cardiomyopathy in the last three years.
|
Saudi Arabia N=25 |
Lebanon
N=24 |
Egypt
N=24 |
Iraq
N=12 |
|
|
Sub-specialties |
||||
|
Heart failure cardiologist |
7 |
8 |
12 |
0 |
|
Imaging cardiologist |
6 |
6 |
2 |
0 |
|
Interventional cardiologist |
12 |
10 |
10 |
12 |
|
All Cardiologists combined |
25 |
24 |
24 |
12 |
|
Experience |
|
|
|
|
|
Average number of heart failure diagnoses per month |
14.7 |
41.9 |
20.8 |
|
|
Average number of ATTR-CM diagnoses in the last 3 years |
13.1 |
3.3 |
8.5 |
0.7 |
|
Average number of AL-CM diagnoses in the last 3 years |
14 |
2.7 |
7.9 |
0.6 |
|
Average number of AA diagnosis the last 3 years |
11.5 |
1.9 |
6.2 |
0.7 |
As for the resources available in their affiliated hospitals, 18% of participants reported having neither heart failure nor cardiomyopathy units, 42% had heart failure units, 2% had cardiomyopathy units, and 38% had both specialty units. Almost 58% reported their affiliated hospital having a heart failure training program, while 93% reported their affiliated hospital as a tertiary-care referral center (Table 2).
|
|
Lebanon |
Iraq |
Saudi Arabia |
Egypt |
Total Average |
|
Heart failure training program |
92% |
0% |
96% |
50% |
68% |
|
Tertiary-care referral center |
92% |
100% |
100% |
83% |
93% |
The availability of electronic medical records was reported to be 71% overall among our participants, being 100% in Saudi Arabia and Iraq, 92% in Lebanon, and 83% in Egypt. The most common diagnostic code (65% of participants) used to track heart failure patients was the International Classification of Diseases (ICD) 10 WHO. Almost 96% used ICD 10 WHO in both Lebanon and Saudi Arabia compared to 33% and 17% in Iraq and Egypt, respectively, who reported using other systems.
Concerning the availability of diagnostic tools, the majority of the participants (86%) reported the availability of cardiac magnetic resonance imaging (MRI); however, 5% reported having none of the diagnostic modalities, while only 45% reported the availability of Echo Bone scintigraphy (Figure 1).
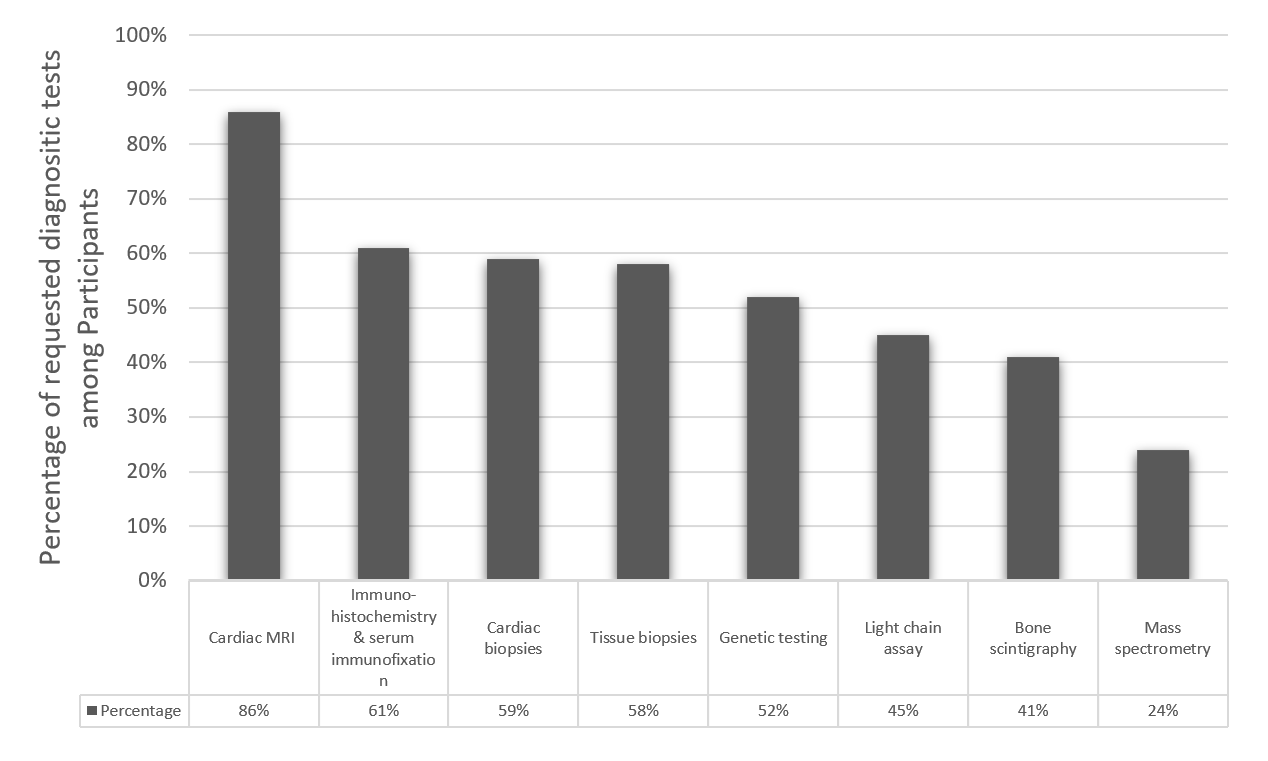
Figure 1. Percentage of participants reporting availability of diagnostic tools.
The mean number of heart failure patients with reduced or preserved EF diagnosed by participating cardiologists in a month was 26, where 45% of HF patients (a mean of 11.7) had heart failure with preserved ejection fraction. Overall, the mean number of patients diagnosed with CA over the last three years was 7.8 for ATTR-CM, 7.8 for AL-CM, and 6.3 for AA-CM (Table 3).
|
|
Lebanon Mean (range) |
Iraq Mean (range) |
Saudi Arabia Mean (range) |
Egypt Mean (range) |
All countries Mean (range) |
|
ATTR-CM |
3.3 (0 – 8) |
0.7 (0 – 2) |
10.5 (4 – 30) |
8.5 (0 – 30) |
6.8 (0 – 30) |
|
AL-CM |
2.7 (0 – 5) |
0.6 (0 – 2) |
11.3 (4 – 22) |
7.9 (0 – 50) |
6.7 (0 – 50) |
|
AA-CM |
1.9 (0 – 5) |
0.7 (0 – 2) |
9.9 (3 – 16) |
6.2 (0 – 30) |
5.7 (0 – 30) |
Beliefs and knowledge of cardiac amyloidosis
The participating physicians were also asked about their knowledge of CA. Around 22% of the participants believed that the heart was affected in 100% of systemic amyloidosis cases, and 33% believed it affects the heart in 60 – 75% and 30 – 40% of the cases, respectively. Surprisingly 12% believed that cardiac involvement is rare in systemic amyloidosis. Regarding the perceived prevalence of each type of amyloidosis, 24% believed that the prevalence of hereditary mutant TTR amyloidosis (ATTRm) is the same worldwide, and 13% believed that the prevalence of wild-type TTR amyloidosis (ATTRwt) is not well known. Almost 24% believed that neither prevalence statements accurately represented the prevalence of ATTR-CM, and 40% believed both statements were accurate. Participants believed that the main causes of CA were as follows: (i) deposit of amyloid fibrils due to mutant or wild-type transthyretin (51%), (ii) deposit of amyloid fibrils due to abnormal monoclonal light chains immunoglobulins (35%), (iii) there are 12 mutations worldwide for the ATTRm type CA (8%), and (iv) there are no other proteins precursors that could give amyloid fibrils (2%).
As per the physicians’ beliefs about symptoms raising suspicion of CA, most physicians consider amyloidosis in case of heart failure symptoms with poor response or intolerance to standard therapy (78%), in concentric left ventricle hypertrophy (71%), and in preserved or mildly reduced ejection fraction (69%). On the other hand, 53% of participants considered moderately or severely reduced ejection fraction as a characteristic raising the suspicion of CA (Figure 2).
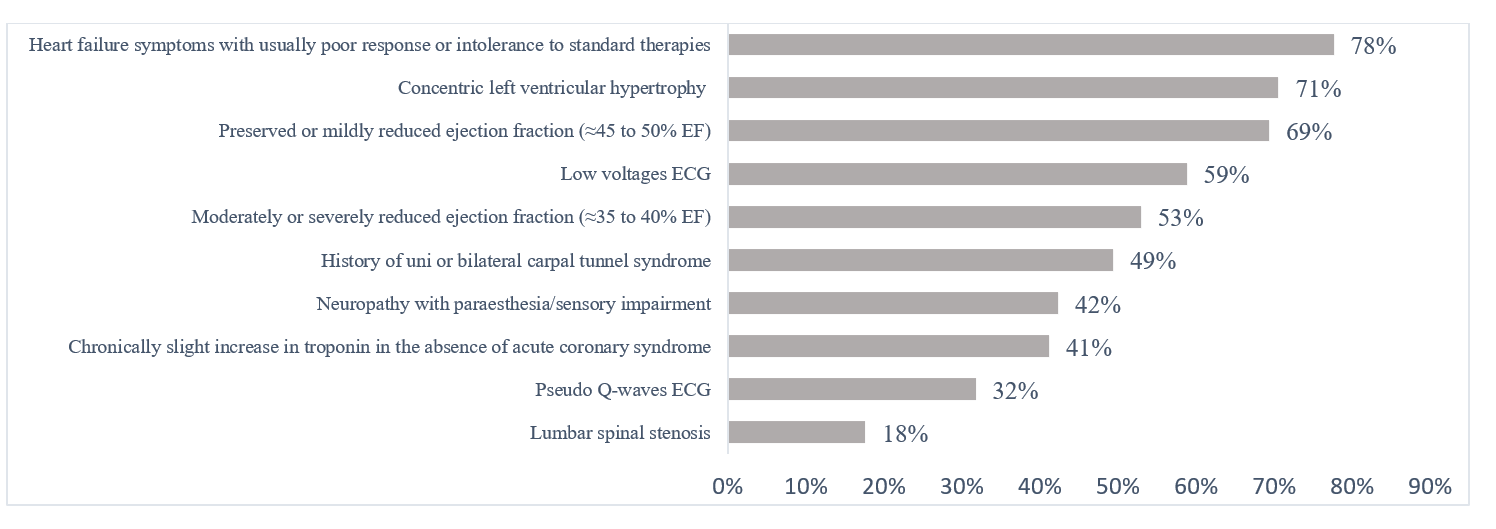
Figure 2. Clinical signs and symptoms raising suspicion of cardiac amyloidosis among the participants.
Practices in diagnosis and treatment
Regarding the perceived misdiagnosis rate of CA, 24% of the participants believed that more than 75% of patients with CA were misdiagnosed, 33% believed that misdiagnosis is among many patients between 51 – 75%, and 29% believed that a moderate number of patients (26-50%) were misdiagnosed. Only 14% believed that less than 25% of patients were misdiagnosed (Table 4).
|
Perception of misdiagnosis of cardiac amyloidosis |
Saudi Arabia N (%) |
Lebanon N (%) |
Egypt N (%) |
Iraq N (%) |
|
>75% of the patients are misdiagnosed |
4 (16) |
6 (25) |
4 (17) |
6 (50) |
|
51-75% of the patients are misdiagnosed |
9 (36) |
9 (38) |
8 (33) |
2 (17) |
|
26-50% of the patients are misdiagnosed |
10 (40) |
4 (17) |
7 (29) |
4 (33) |
|
<25% of the patients are misdiagnosed |
2 (8) |
5 (21) |
5 (17) |
0 (0) |
Regarding the clinical practices of the participants, 39% consider cardiac MRI as the first imaging test order for suspected CA cases, while surprisingly, only 10% would consider a myocardial scintigraphy scan. Most (82%) of the participating physicians would request urine protein immune-electrophoresis (UPEP) or (75%) serum protein immune-electrophoresis (SPEP) to rule out systemic light-chain amyloidosis. Other requested tests are mentioned in (Table 5).
|
Diagnostic test |
All physicians (n=85) |
|
A. First Imaging Test for Suspected Cardiac Amyloidosis |
|
|
Cardiac MRI |
39% |
|
Echocardiogram |
33% |
|
Cardiac CT |
18% |
|
Myocardial scintigraphy scan |
11% |
|
B. Biological Tests to Rule Out Systemic Light Chain Amyloidosis |
|
|
UPEP |
82% |
|
SPEP |
75% |
|
Kappa and Lambda Free light chains ratio |
62% |
|
NT-ProBNP and Troponin |
22% |
|
CT: Computerized Tomography; MRI: Magnetic Resonance Imaging; NT-ProBNP: Natriuretic peptide tests measure levels of BNP; SPEP: Serum Protein Electrophoresis; UPEP: Urine Protein Electrophoresis |
|
When considering all diagnostic modalities, the top go-to diagnostic test overall for CA for the participants is echocardiogram (92%), cardiac biopsy (78%), genetic testing (58%), free light chain assay (51%), SPEP/UPEP (49%), non-cardiac tissue biopsy (49%), and bone scintigraphy (40%). In addition, almost 2/3 (74%) of the physicians believed a cardiac biopsy is mandatory to confirm ATTR-CM, and 82% voted for a peripheral tissue biopsy to confirm the presence of light chain amyloidosis. As for genetic testing, 27% considered it necessary for the diagnosis, 21% considered it helpful but not necessary, 2% not necessary, and 47% believed that necessity depends on each case.
Regarding treatment options, only 53% of the participants believed that a difference exists in the treatment of ATTR-CM and light chain amyloidosis; 26% of the participants didn’t believe that there was a difference in treatment, and 21% were not sure about the treatment modality. Regarding potential treatment options for ATTR-CM, 21% did not consider proteasomes inhibitors as a potential treatment. The majority (40%) believed that tetramer stabilizers, fibrils disrupters, proteasomes inhibitors, and RNA silencers were not potential options to treat ATTR-CM.
Need for and interest in education
The majority of physicians reported the need for more information on the diagnosis and treatment of CA, with 76% of participants reporting high interest in learning about current and future treatments for ATTR-CM. In addition, 75% of the physicians believed it would be highly beneficial to link with international experts through telemedicine platforms to diagnose ATTR-CM accurately, and 69% of them endorsed the high benefit of networking with professionals from neighboring countries with approved ATTR-CM products and learning from their experience. The highest rates of interest in virtual contact with international experts or networking with neighboring countries were in Saudi Arabia, followed by Lebanon, Iraq, and Egypt.
Discussion
The disease knowledge of physicians in the field of CA is a continuous area of interest. With the increasing incidence and prevalence of CA, there is a need to ensure the proper understanding and correct misbeliefs related to the patient journey from diagnosis to management [13]. This study aimed to understand better the current beliefs and knowledge of CA, including the diagnostic tools and management, and the interest and need for relevant education. There was a clear variation among the participants in the knowledge of cardiac amyloidosis diagnosis and management. The level of knowledge and engagement with CA was higher in countries like Saudi Arabia and Lebanon. The variation in the experience level was seen in other populations, where among physicians responding to a similar study in Switzerland, 15% considered their knowledge to be of expert level. The study has shed light on the awareness and mastery of CA disease among physicians in the selected countries.
In general, participants involved in the survey have encountered a relatively small number of cases with CA. The presence of electronic medical records in most cardiology centers in the region could be an encouraging research potential [14,15]. There is still a need to unify the classification of diseases in some countries in the region. The vast majority of Saudi and Lebanese physicians use ICD-10 WHO in cardiology-related research.
Although most of the physicians engaged in this survey worked in tertiary-care referral centers, there was still a need for more specialty units related to heart failure and cardiomyopathy. In addition, there is a clear need for heart failure training programs in the region. A minimal number of physicians had no available diagnostic modalities for CA in their centers. This could be due to their presence in rural areas or being in non-tertiary cardiology centers. However, decentralization of cardiology centers with diagnostic and management modalities is still required in the region, especially with the wide geographic distribution in four countries. Regarding the diagnostic modalities, a significant proportion of the participants believe that invasive procedures like cardiac biopsy are needed to diagnose cardiac amyloidosis. While cardiac biopsy is still perceived, as per the results of this survey, to be the gold standard diagnostic tool, the current recommendations limit its use to specific cases after the advances in cardiac imaging techniques, notably the cardiac radionuclide scintigraphy [16].
CA is caused by the progressive infiltration of amyloid fibrils composed of either monoclonal immunoglobulin light chains (AL) or transthyretin (ATTR). In addition, ATTR is either hereditary or acquired. Hereditary ATTR (ATTRm) results from a mutation in the genetic coding of the TTR protein with more than 70 identified transthyretin mutations [16]. A Saudi study analyzed 13,905 Saudi exomes of unrelated populations. Three novel TTR mutations were discovered, in addition to three known TTR variants [6]. This sheds light on the importance of a mass genetic screening of individuals with family history to identify the patients who are at high risk. However, acquired ATTR is believed to be due to age-related protein misfolding. Participants from our region had a fair understanding of the pathophysiology of CA. A combination of compatible cardiac findings and the systemic involvement of multiple organs such as the kidney, CNS, or plasma cells should raise the suspicion index for CA. More than half of the participants in our survey believed that cardiac involvement occurs in most cases with systemic amyloidosis. This misbelief could result from the increasing prevalence and recognition of ATTR-CM by cardiologists [17].
ATTR-CM is relatively rare, with heterogeneous symptoms that could be mistaken for other more common diseases. The latest ESC guidelines highlighted the cardiac and extracardiac red flags to help suspect CA [18]. Most participating physicians in this study suspected CA in patients with heart failure symptoms not responding to standard therapy. On cardiac imaging, left ventricular hypertrophy and mild reduction in ejection fraction raised the suspicion of CA. Similarly, 75% of physicians participating in the Swiss study believed that left ventricular hypertrophy with normal blood pressure is the main indicator of CA [19]. Similar to our study, absolute low-voltage ECG was the main ECG finding to suspect CA; however, 88% of the Swiss group believed this versus 59% in our group. However, previous literature indicated that absolute low QRS voltages are not a common ECG finding, especially in ATTR-CM and studies have recently focused more on relative low voltage as it is more frequent in ATTR-CM compared to absolute low ECG voltage [20,21].
Although our participants had a fair knowledge of symptoms and signs of CA, there is still room for more awareness of the red flags of ATTR-CM. Hypertrophic cardiomyopathy is a common misdiagnosis that needs to be ruled out by signs such as asymmetrically increased septal wall thickness and other diagnostic findings using bone scintigraphy, cardiac MRI, and genetic testing [17,22]. The perceptions of our participants widely varied regarding the rate of CA misdiagnosis; however, more than half of our participants believed there was a high rate of misdiagnosis. This could be due to several factors, including low disease suspicion index, fragmented information about the disease, clinical presentation of CA that could be variable from one patient to another, and lack of experience in CA. A previous study observed that the diagnosis of 42% of ATTRwt was delayed for more than four years [9]. An Egyptian study discovered that 1 out of 11 patients with undiagnosed cardiomyopathy had CA when undergoing cardiac biopsy [23]. Hence, it’s critical not to delay or falsely label the diagnosis of CA. Accordingly, non-invasive diagnostic modalities should be recommended in patients with cardiomyopathy and heart failure of unknown origin.
Surprisingly, a considerable number of participants were unaware of the role of bone scintigraphy of the heart in diagnosing CA. This awareness level was similar to the Swiss survey results, where only 44% would arrange for it in their routine investigation [14]. According to ESC recommendations, bone scintigraphy of the heart was proven to have excellent specificity and sensitivity for ATTR-CM as a non-invasive technique [24] and is considered an essential confirmatory test for ATTR-CA in the absence of AL in patients with clinical, echocardiographic and electrocardiographic ‘red flags’ for CA [25]. The low awareness level of this diagnostic tool could explain the low diagnosis rate of ATTR-CM. National and regional efforts should be exerted to make this modality widely known and available in cardiology centers and raise awareness of its importance and indications. Additionally, training should be done for nuclear medicine specialists on the correct protocol to better use this imaging modality and how to interpret the results to avoid any false diagnosis.
On the other hand, echocardiography and cardiac biopsy were among the highest “go-to” diagnostic modalities. In addition, cardiac and tissue biopsies were believed to confirm ATTR-CM and light chain amyloidosis, respectively. However, a cardiac biopsy is not a common practice in our region and is considered an invasive intervention with a significant risk of bleeding, arrhythmia, and perforation. Although cardiac biopsy may have the gold standard for diagnosing ATTR-CM, previous evidence demonstrated that grade 2-3 uptake on bone scintigraphy of the heart has a 100 % positive predictive value for ATTR-CM after exclusion of AL using monoclonal protein biomarkers [26]. The latest ESC guidelines highlighted the importance of serum and urine protein electrophoresis in detecting monoclonal proteins; this will support the screening of ATTR-CM in family members in case of identified mutant type [25]. NT-ProBNP and troponin were among the lowest biological tests cited in our survey to rule out cardiac involvement, suggesting a lack of deep knowledge of CA. Previous studies supported that these two biomarkers could be elevated with cardiac impairment in both AL and ATTR [27].
Most participants from the study centers believed that genetic testing should be individualized among patients with ATTR-CM. However, the ESC guidelines specifically recommended assessing ATTR mutation status in patients with grade 2 or 3 cardiac uptake on scintigraphy and negative monoclonal proteins [26]. Accordingly, genetic testing is essential in all ATTR-CM patients, irrespective of age. Also, screening family members in case of hereditary type is recommended, which will support an early diagnosis of the diseases once symptoms appear. In addition, there is limited availability of this diagnostic modality in main cardiology centers in the four selected countries.
There is a large gap in ATTR-CM management knowledge among the participants in our region. A considerable number of our participants believed that the management of ATTR-CM and light-chain CM are essentially the same. A recent study observed that significantly different heart failure courses exist between the two types of cardiomyopathies, including hospital admissions due to heart failure (HR 2.87, p = 0.003) and mortality (HR 2.51, p = 0.015) [28]. In addition, it is proven that AL carries the worst prognosis among cardiac amyloidosis and is considered a medical emergency to treat the underlying primary malignant cause of the disease [29]. Misconceptions, such as the role of proteasome inhibitors in ATTR-CM, highlighted the need for proper education and knowledge exchange. Proteasomes inhibitors, such as bortezomib, have a role in managing AL amyloidosis; however, there is no evidence or previous randomized controlled studies on their role in ATTR-CM. Tetramer stabilizer molecules (tafamidis) and genetic silencers (patisiran and inotersen) are potential treatment options specific for ATTR-CM [30]. In addition, tafamidis is the cornerstone of ATTR-CM management and the only drug that showed a significant benefit in both ATTRm and ATTRwt [25]. A phase-III randomized control trial showed that ATTR-CM patients receiving tafamidis had lower hospitalization rates and all-cause mortality (p<0.001) compared to patients receiving placebo [30]. Several factors may impact this knowledge level, including drug availability, lack of continuous medical education in this disease area, limited knowledge exchange with international experts, and minimal local experts in CA.
The survey highlighted the willingness and interest of most physicians to understand the proper management paradigm. Fortunately, most participants showed a high interest in more training, collaboration with international experts, and networking with neighboring countries, with some differences in the rates between participants based on their countries of affiliation. The majority of the participants endorsed teleconferences and collaborations with international and regional experts in ATTR-CM. One major outcome of this study is to increase awareness about CA, highlight the need for more specialists in the field, and push for more training and educational conferences for cardiologists to keep CA on their differential diagnosis list. Hence cardiologists should be aware of the major signs and red flags that could indicate the presence of CA.
To our knowledge, this is the first study that analyzes the awareness of CA among cardiologists in Arab countries. Nonetheless, limitations do exist in the study. The most notable was the small sample size of 85 physicians who completed the survey compared to the number of practicing cardiologists in the four countries. In addition, only four Middle Eastern countries were selected for this study leading to important limitations and selection bias. Thus, the results should be interpreted this way and may not be generalized to all countries, especially with the limited sample size. Further large-scale studies are needed to accurately map the CA knowledge and awareness in the Middle Eastern world.
Conclusion
Surveying cardiologists in four selected Middle Eastern countries (Saudi Arabia, Lebanon, Egypt and Iraq) has highlighted significant knowledge disparities and gaps in the diagnosis, management, and treatment of ATTR-CM. Physicians from the corresponding countries will likely benefit from additional training and exposure to the latest advances in managing this disease. These measures would be beneficial to ensure better patient care and lower rates of late referrals or misdiagnosis.
Conflict of Interest
IA and MG are Pfizer employees and were involved in study conception and manuscript approval. All other authors report no conflict of interest.
Funding
This study was sponsored by Pfizer.
Acknowledgment
The data generated and analyzed in this study is available with the corresponding author upon request. We’d like to thank CTI MEA for their support in medical writing and editing support.
Author Contributions Statement
All authors contributed to concept and design, literature search, data collection, analysis and interpretation of results, manuscript preparation, manuscript editing, and manuscript review. This manuscript has been read and approved by all the authors.
References
2. Esplin BL, Gertz MA. Current Trends in Diagnosis and Management of Cardiac Amyloidosis. Current Problems in Cardiology. 2013;38(2):53-96.
3. Siddiqi OK, Ruberg FL. Cardiac amyloidosis: An update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med. 2017/07/13. 2018 Jan;28(1):10-21.
4. Kyriakou P, Mouselimis D, Tsarouchas A, Rigopoulos A, Bakogiannis C, Noutsias M, et al. Diagnosis of cardiac amyloidosis: a systematic review on the role of imaging and biomarkers. BMC Cardiovasc Disord. 2018 December 4;18(1):221.
5. Mohty D, Damy T, Cosnay P, Echahidi N, Casset-Senon D, Virot P, et al. Cardiac amyloidosis: Updates in diagnosis and management. Archives of Cardiovascular Diseases. 2013;106(10):528-40.
6. Abouelhoda M, Mohty D, Alayary I, Meyer BF, Arold ST, Fadel BM, et al. Established and candidate transthyretin amyloidosis variants identified in the Saudi population by data mining. Hum Genomics. 2021 August 11;15(1):52.
7. Brunjes DL, Castano A, Clemons A, Rubin J, Maurer MS. Transthyretin Cardiac Amyloidosis in Older Americans. J Card Fail. 2016/10/18. 2016 Dec;22(12):996-1003.
8. Sharma N, Howlett J. Current state of cardiac amyloidosis. Current Opinion in Cardiology. 2013;28(2):242-8.
9. Lane T, Fontana M, Martinez-Naharro A, Quarta CC, Whelan CJ, Petrie A, et al. Natural History, Quality of Life, and Outcome in Cardiac Transthyretin Amyloidosis. Circulation. 2019 July 2;140(1):16-26.
10. Bishop E, Brown EE, Fajardo J, Barouch LA, Judge DP, Halushka MK. Seven factors predict a delayed diagnosis of cardiac amyloidosis. Amyloid. 2018 July 3;25(3):174-9.
11. Rozenbaum MH, Large S, Bhambri R, Stewart M, Whelan J, van Doornewaard A, et al. Impact of Delayed Diagnosis and Misdiagnosis for Patients with Transthyretin Amyloid Cardiomyopathy (ATTR-CM): A Targeted Literature Review. Cardiol Ther. 2021/04/20. 2021 Jun;10(1):141-59.
12. Ladefoged B, Dybro A, Povlsen JA, Vase H, Clemmensen TS, Poulsen SH. Diagnostic delay in wild type transthyretin cardiac amyloidosis – A clinical challenge. International Journal of Cardiology. 2020 April 1;304:138-43.
13. Nguyen FD, Rodriguez M, Krittanawong C, Witteles R, Lenihan DJ. Misconceptions and Facts About Cardiac Amyloidosis. American Journal of Cardiology. 2021 December 1;160:99-105.
14. Cowie MR, Blomster JI, Curtis LH, Duclaux S, Ford I, Fritz F, et al. Electronic health records to facilitate clinical research. Clin Res Cardiol. 2016/08/24. 2017 Jan;106(1):1-9.
15. Farsi M al, West DJ. Use of Electronic Medical Records in Oman and Physician Satisfaction. Journal of Medical Systems. 2006;30(1):17-22.
16. Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing common questions encountered in the diagnosis and management of cardiac amyloidosis. Circulation. 2017 Apr 4;135(14):1357-77.
17. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019 June 11;73(22):2872-91.
18. Garcia-Pavia P, Rapezzi C, Adler Y, Arad M, Basso C, Brucato A, et al. diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2021;42(16):1554-68.
19. Mircsof D. Diagnosis of Amyloidosis: A Survey of Current Awareness and Clinical Challenges Among Cardiologists in Switzerland. Cardiology and Therapy. 2020;9(1):127-38.
20. Rapezzi C, Lorenzini M, Longhi S, Milandri A, Gagliardi C, Bartolomei I, et al. Cardiac amyloidosis: the great pretender. Heart Failure Reviews. 2015;20(2):117-24.
21. González-López E, Gagliardi C, Dominguez F, Quarta CC, de Haro-del Moral FJ, Milandri A, et al. Clinical characteristics of wild-type transthyretin cardiac amyloidosis: disproving myths. European Heart Journal. 2017 Jun 21;38(24):1895-904.
22. Brenes JC, Doltra A, Prat S. Cardiac magnetic resonance imaging in the evaluation of patients with hypertrophic cardiomyopathy. Glob Cardiol Sci Pract. 2018 August 12;2018(3):22.
23. Hassan AKM, Fouad DA, Refaiy A. Demographic features and prevalence of myocarditis in patients undergoing transarterial endomyocardial biopsy for unexplained cardiomyopathy. Egypt Heart J. 2016/10/10. 2017 Mar;69(1):29-35.
24. Treglia G, Glaudemans AWJM, Bertagna F, Hazenberg BPC, Erba PA, Giubbini R, et al. Diagnostic accuracy of bone scintigraphy in the assessment of cardiac transthyretin-related amyloidosis: a bivariate meta-analysis. European Journal of Nuclear Medicine and Molecular Imaging. 2018;45(11):1945-55.
25. Garcia-Pavia P, Rapezzi C, Adler Y, Arad M, Basso C, Brucato A, et al. diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. European Heart Journal. 2021 April 21;42(16):1554-68.
26. Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation. 2016 June 14;133(24):2404-12.
27. Damy T, Deux J-F, Moutereau S, Guendouz S, Mohty D, Rappeneau S, et al. Role of natriuretic peptide to predict cardiac abnormalities in patients with hereditary transthyretin amyloidosis. Amyloid. 2013 December 1;20(4):212-20.
28. Itzhaki Ben Zadok O, Vaturi M, Vaxman I, Iakobishvili Z, Rhurman-Shahar N, Kornowski R, et al. Differences in the characteristics and contemporary cardiac outcomes of patients with light-chain versus transthyretin cardiac amyloidosis. PLoS One. 2021 Aug 9;16(8):e0255487-e0255487.
29. Falk RH, Alexander KM, Liao R, Dorbala S. AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy. J Am Coll Cardiol. 2016 Sep 20;68(12):1323-41.
30. Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. New England Journal of Medicine. 2018 August 27;379(11):1007-16.