Abstract
Bannayan-Zonana syndrome (BRRS) is a rare genetic disorder characterized by macrocephaly, numerous soft tissue and visceral hamartomas, and lipomas. Because of the risk of fatal bleeding and visceral neoplasia in adulthood, recognizing this disease is critical. We reported a new pediatric case of BRRS in an 18-month-old female infant with lipomas, macrocephaly, and gastrointestinal hamartomatous polyps that prompted ileo-ileal intussusception and protein-losing enteropathy. Patients with BRRS require a comprehensive approach. Our case is unique in its sporadic occurrence and the presence of protein-losing enteropathy as a gastrointestinal polyposis manifestation. Pediatric patients with multiple lipomas, protein-losing enteropathy, and intestinal intussusception should be assessed for BRRS.
Keywords
Bannayan-Zonana syndrome, Polyposis, Intussusception, Protein losing enteropathy, Child
Introduction
Bannayan-Zonana syndrome, also known as Bannayan-Riley-Ruvalcaba syndrome (BRRS), is a rare genetic disease characterized by macrocephaly with various soft tissue and visceral hamartomas and lipomas [1]. Mutations in the PTEN gene cause BRRS. The inheritance pattern is autosomal dominant, with a few sporadic occurrences documented [2-4]. BRRS's prevalence is currently unclear. The danger of fatal hemorrhages and visceral neoplasia in adulthood necessitates knowledge of this condition. We discuss the case of an 18-month-old female infant with BRRS who also had subcutaneous lipomas, macrocephaly, and extensive intestinal polyposis, as well as ileo-ileal intussusception and protein-losing enteropathy.
Case Report
An 18-month-old female girl presented with generalized edema, dyspnea, and bilateral cervical and axillary masses. She was the third child of non-consanguineous parents. She was born at term after a spontaneous delivery. The birth weight was 3300 g (50th percentile), the head circumference was 35 cm (75th percentile), and the birth length was 50 cm (50th percentile). Her family history was significant for multiple lipomas in one sibling (her grandmother). At the age of eight months, she was hospitalized in the dermatology department to explore cervical and axillary tumors. A clinical examination revealed a multiple elastic soft subcutaneous tumor with a diameter of 12 cm that affected the neck and thorax. Ultrasonography was performed, and the lesions were diagnosed as lipomas, but a biopsy wasn’t performed. There was no history of associated gastro-intestinal disorders. The evolution was marked by the increase in size of the soft, elastic subcutaneous tumors and the appearance of dyspnea and generalized edema.
Physical examination revealed that the weight was 10.5 kg (+0.5 DS), the height was 80 cm (+2 DS), and the head circumference was 51 cm (+2 DS). She had two painless, elastic soft subcutaneous tumors on her neck, linked on the median line, and associated with a left axillary mass (Figure 1). All of the tumors were solid, retentive, and suggestive of lipomas; others were small and located in the cephalic area. There was no thrill on palpation in favor of their vascular character. The child also had generalized soft white edema, that allowed the cup to remain on the child's face and limbs. The neurological examination was unremarkable.
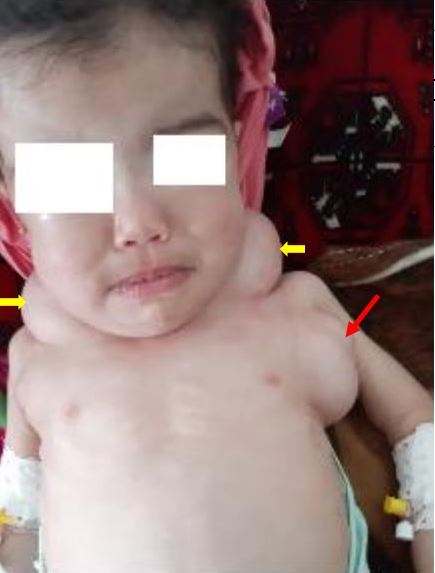
Figure 1. The child presented with multiple soft-consistency masses affecting the neck and thorax (arrows).
A complete blood cell count and laboratory tests revealed severe anemia (Hb: 7 g/dl, VGM: 56,5 fl, CCMH: 25) and hypoproteinemia at 29 g/l. The concentration of alpha-1-antitrypsin in a stool sample was increased (>3600 g/g). Therefore, we performed a thyroid ultrasonography and thyroid hormone dosage, which were both normal. Thyroid antibodies were also negative. Cranial tomography was normal. A computed tomography (CT) scan was performed to better understand the nature and extension of the masses. It revealed an extensive lipomatosis in the thorax-neck region and showed a thickening of the bowel wall with intussusception diagnosed accidentally in the left lower quadrant (Figure 2).
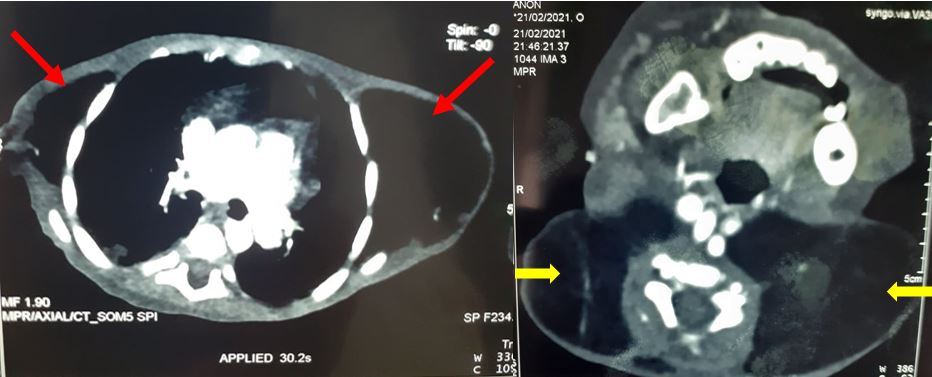
Figure 2. CT scan images revealing an extensive lipomatosis in the thorax and neck region (arrows).
An exploratory laparotomy was performed, and four ileo-ileal intussusceptions measuring 15 cm were discovered (Figure 3). Manual reduction was performed. The invaginated segment was infiltrated and thickened, so we opted for resection of the involved segment and an end-to-end anastomosis. Histopathological examination confirmed the diagnosis of intestinal hamartomatous polyps (Figure 4). One tumor in the left armpit measuring 6x8 cm was excised and was pathologically diagnosed as a lipoma (Figure 5).
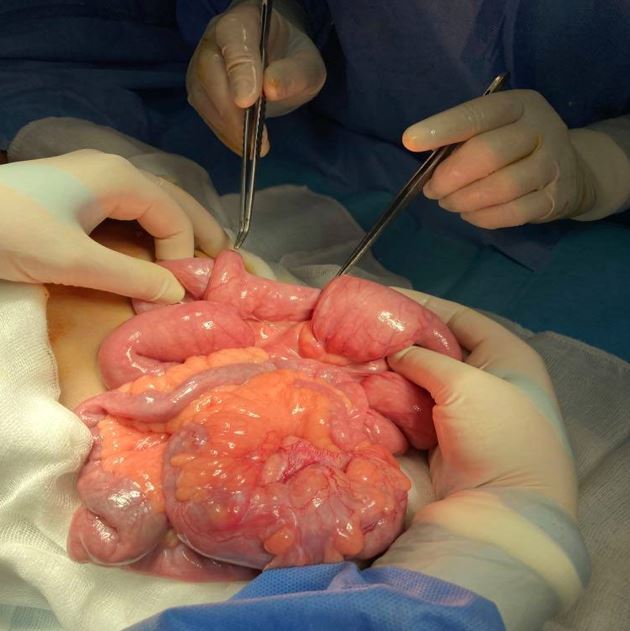
Figure 3. Preoperative view of ileo-ileal intussusceptions.
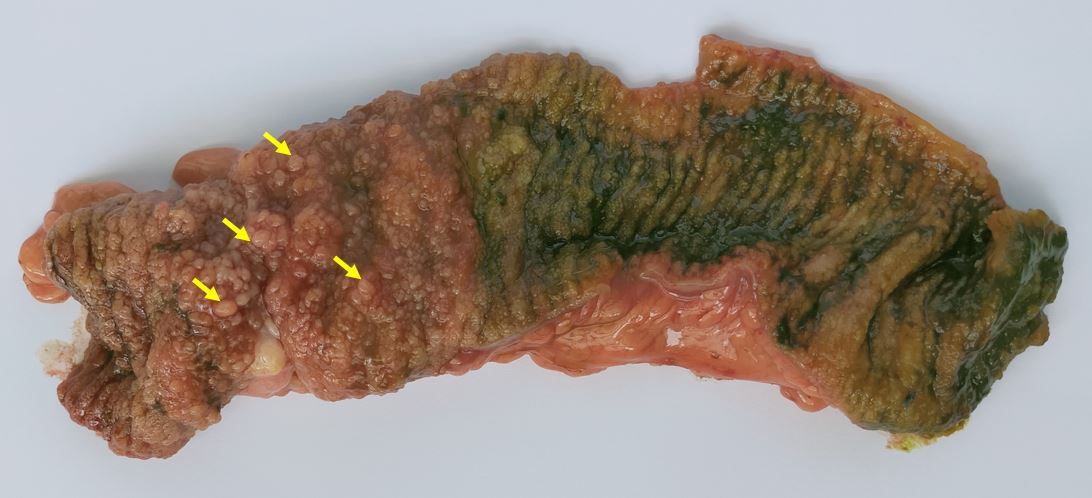
Figure 4. The macroscopic appearance of the resected specimen revealed diffuse polypoid lesions projecting into the lumen. Yellow arrows highlight some of the polyps.
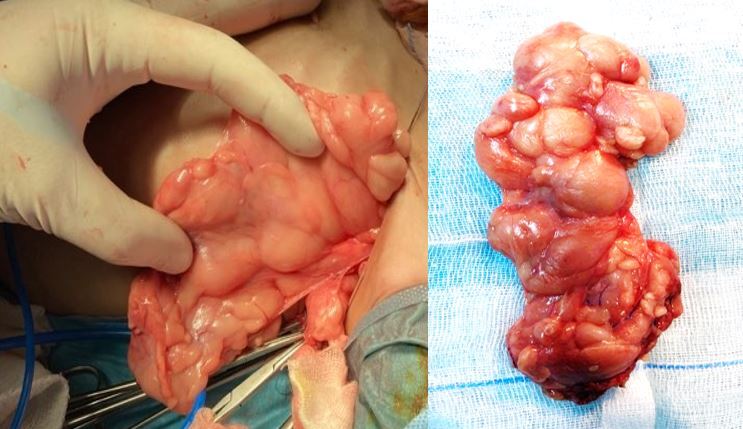
Figure 5. Perioperative view of the tumor located in the left armpit, which was excised and pathologically diagnosed as a lipoma.
The postoperative course was uneventful for this child. She has been followed up regularly, and she is currently doing well, one year after the diagnosis. Molecular analysis of the PTEN gene wasn’t available in our country.
Discussion
BRRS is characterized by congenital macrocephaly (>2 standard deviations [SD]), the presence of lentigines or pigmented maculae on the penis, hamartomatous intestinal polyps, arteriovenous malformations, autism, developmental delay, or intellectual disability, and lipomas [2].
Currently, there is still no consensus criterion for the diagnosis of BRRS. Although the syndrome is usually diagnosed if at least 3 of the 4 major features (macrocephaly, lipomatosis, hemangiomas, and speckled penis) are found, according to Marsh and Laury et al. [5,6]. In some cases, the presence of two features is considered sufficient [7]. In our case, BRRS was diagnosed based on the observation of two features (macrocephaly and hamartomas).
Frontal bossing, hypertelorism, downward sloping palpebral fissures, ogival palate, strabismus, epicanthus inversus long philtrum, a thin upper lip, and relative micrognathia are examples of facial dysmorphic features [8].
Mild neurological dysfunction, growth, speech and motor delays, mental retardation, and seizures have also been reported [9].
A wide variety of thyroid disorders have been described in children with BBRS. These include benign and malignant lesions and autoimmune conditions. Benign disorders include nodular goiter, single, or multiple follicular adenomas, autoimmune thyroid disease including Hashimoto's thyroiditis and lymphocytic thyroiditis. The risk of thyroid cancer in adulthood is reported to be increased above the general population risks [2]. Hence, our patient underwent thyroid ultrasonography which was normal; thyroid hormone dosage was also normal and thyroid antibodies were negative.
GI tract involvement is seen in 35–45% of the patients; polyps may be seen throughout the GI tract but generally occur in the lower GI tract and are reported as hamartomatous [8,9]. GI polyposis may cause chronic bleeding, anemia, intussusception, and diarrhea. In our case, diffuse polyposis was responsible for anemia, intussusception, and protein-losing enteropathy. This is, to our knowledge, the first pediatric case of protein-losing enteropathy associated with multiple ileo-ileal intussusception in BRRS.
Inheritance is by autosomal dominant transmission; however, sporadic cases have been reported [1,3,4]. BRRS is considered part of the PTEN hamartoma tumor syndrome (PHTS) [4,5,9]. Molecular analysis of the PTEN gene wasn’t available in our country. Our case seems to be sporadic. The PTEN gene is a tumor suppressor gene found on chromosome 10q23.3. Mutations of this gene are detected in 60% of the patients with BRRS. It leads to the unregulated cellular proliferation of three embryogenic germ layer cells. As a result, ectodermal, mesodermal, and endodermal hamartomas may evolve in this group of patients.
Many reviews in the literature focus on PHTS as an adult hamartoma and malignancy predisposition condition [5-9]. As a result, patients with BRRS may develop different malignancies in adulthood, such as GI adenocarcinoma; thus, they require annual surveillance beginning in childhood, with hemoglobin level and fecal occult blood test [1]. In addition to this, periodic surveillance of other related tumors, such as breast, thyroid, endometrial, skin, and renal cancers, is indicated in PTEN mutation-positive BRRS [1].
Conclusion
Patients diagnosed with BRRS need a multidisciplinary approach. Our case is original in its sporadic occurrence and the presence of protein-losing enteropathy as a manifestation of GI polyposis. BRRS should be considered in pediatric patients with multiple lipomas, protein-losing enteropathy, and intestinal intussusception.
Acknowledgements
None.
Authors' Contributions
IM: Data analysis, draft the manuscript, revised the manuscript and approved the manuscript. MBD: operated the patient, manuscript writing and literature review. TC: Conception, data analysis, draft the manuscript, revised the manuscript, and approved the submission. MZ: Acquisition and analysis, revised the manuscript, and approved the manuscript. All authors have read and approved the manuscript.
References
2. Gujrati M, Thomas C, Zelby A, Jensen E, Lee JM. Bannayan-zonana syndrome: a rare autosomal dominant syndrome with multiple lipomas and hemangiomas: A case report and review of literature. Surg Neurol. 1998 August;50(2):164?8.
3. Lynch NE, Lynch SA, McMenamin J, Webb D. Bannayan–Riley–Ruvalcaba syndrome: a cause of extreme macrocephaly and neurodevelopmental delay. Arch Dis Child. 2009;94(7):553‑4.
4. Blumenthal GM, Dennis PA. PTEN hamartoma tumor syndromes. Eur J Hum Genet. 2008;16(11):1289?300.
5. Marsh DJ, Kum JB, Lunetta KL, Bennett MJ, Gorlin RJ, Ahmed SF, et al. PTEN Mutation Spectrum and Genotype-Phenotype Correlations in Bannayan-Riley-Ruvalcaba Syndrome Suggest a Single Entity With Cowden Syndrome. Hum Mol Genet. 1999;8(8):1461?72.
6. Laury AR, Bongiovanni M, Tille J-C, Kozakewich H, Nosé V. Thyroid Pathology in PTEN-Hamartoma Tumor Syndrome: Characteristic Findings of a Distinct Entity. Thyroid. 2011;21(2):135?44.
7. Parisi M, Dinulos MB, Leppig K, Sybert V, Eng C, Hudgins L. The spectrum and evolution of phenotypic findings in PTEN mutation positive cases of Bannayan-Riley-Ruvalcaba syndrome. J Med Genet. 2001;38(1):52?8.
8. Latiff ZA, Atmawidjaja RW, RajaLope RJ, Syed Omar SA, Syed Zakaria SZ, Jamal RA. Bannayan Riley Ruvalcaba syndrome. Ann Acad Med Singapore. 2010;39(7):578?572.
9. Edmondson AC, Kalish JM. Overgrowth Syndromes. J Pediatr Genet. 2015 ;04(3):136?43.