Abstract
In the realm of DNA repair, base excision repair (BER) protein, APE1/Ref-1 (Apurinic/Apyrimidinic Endonuclease 1/Redox Effector - 1, also called APE1) has been studied for decades. However, over the past decade, APE1 has been established as a key player in reduction-oxidation (redox) signaling. In the review by Caston et al. (The multifunctional APE1 DNA repair-redox signaling protein as a drug target in human disease), multiple roles of APE1 in cancer and other diseases are summarized. In this Review, we aim to expand on the contributions of APE1 to various diseases and its effect on disease progression. In the scope of cancer, more recent roles for APE1 have been identified in cancer cell metabolism, as well as chemotherapy-induced peripheral neuropathy (CIPN) and inflammation. Outside of cancer, APE1 signaling may be a critical factor in inflammatory bowel disease (IBD) and is also an emergent area of investigation in retinal ocular diseases. The ability of APE1 to regulate multiple transcription factors (TFs) and therefore multiple pathways that have implications outside of cancer, makes it a particularly unique and enticing target. We discuss APE1 redox inhibitors as a means of studying and potentially combating these diseases. Lastly, we examine the role of APE1 in RNA metabolism. Overall, this article builds on our previous review to elaborate on the roles and conceivable regulation of important pathways by APE1 in multiple diseases.
Keywords
Redox effector factor 1, Apurinic/apyrimidinic endonuclease, Redox signaling, Inflammation, Metabolism, Glycolysis, TCA cycle, OXPHOS, Chemotherapy-induced peripheral neuropathy, IBD, Crohn’s, Colitis
Introduction
APE1/Ref-1 (also called APE1) has been extensively studied, initially for its singular role as an endonuclease in DNA base excision repair (BER). However, since the early 2000’s, it has been studied for other roles that have been discovered in addition to DNA repair which include its major role as a reduction-oxidation (redox) signaling protein and its interactions with RNA [1]. Multiple studies report the many functions of APE1/Ref-1 in regulation of key biological functions that control redox homeostatsis [2-4], mitochondrial metabolism [5,6], inflammatory responses [7], neo-vascularization [8,9] and others that make it an attractive target in pathologies such as cancer, chemotherapy-induced peripheral neuropathy (CIPN), inflammatory bowel disease (IBD), retinal ocular diseases [e.g. diabetic retinopathy (DR), diabetic macular edema (DME), and wet age-related macular degeneration (Neovascular AMD)]. APE1’s functional roles are also associated with its localization either within the nucleus or cytoplasm (endoplasmic reticulum or mitochondria) [10,11]. Aditionally, secreted APE1 is reported to induce proinflammatory cytokine IL-6 [12]. APE1 serum levels serve as biomarkers for bladder cancer, hepatocellular carcinoma and oral squamous cell carcinoma [13-16].
APE1’s endonuclease role controlling DNA and RNA metabolism and its redox role regulating transcription factors are attractive targets and so far, several small molecule inhibitors have been developed and are successful in the pre-clinical setting. Only recently, APX3330, an APE1 redox function inhibitor completed Phase I clinical trial with a good safety profile, verified target engagement and a recommended phase II dose (600 mg/d). The review by Caston et al. (The multifunctional APE1 DNA repair-redox signaling protein as a drug target in human disease), focused mostly around APE1’s role in cancer and the use of inhibitors of the redox signaling function of APE1. In this article, we will address some of the less discussed or understudied roles of APE1 that are gaining more recent attention.
APE1 and Cancer Cell Metabolism: New Discoveries
Cancer cells can adapt to meet their ever-changing bioenergetic needs by reprogramming their metabolic pathways thereby acquiring and maintaining unrestricted proliferative capability. Cancer cell metabolic adaptability might be possible due to accumulation of specific mitochondrial metabolites like fumarate, succinate and α-ketoglutarate [17]. To meet their constant demand for fuel especially under nutrient-deprived stress, it is likely that certain malignant cancers shift from glycolysis back to oxidative phosphorylation (OXPHOS) as the main energy supplier [18]. This bidirectional shift might be possible because of mitochondrial metabolic plasticity. In oncogene mutated cancers like KRASG12D-driven PDAC (Pancreatic Ductal Adenocarcinoma) and MYC/KRAS or MYC/ ERBB2-ablated breast cancer, cancer cells predominantly rely on OXPHOS for energy production [17]. Consequently, a previous notion that the Warburg effect is responsible for all tumor survival and growth proves inaccurate due to this dynamic interplay between oxidative metabolism and glycolysis.
The redox signaling activity of APE1 is responsible for facilitating the DNA binding of critical TFs like STAT3 (Signal transducer and activator of transcription 3), NFκB (Nuclear Factor kappa-light chain enhancer of activated B cells), AP-1 (Activator Protein 1) and HIF1α (Hypoxia inducible factor 1 subunit alpha) among others (Figure 1) [19]. APE1 reduces HIF1α thereby activating its DNA binding capability [20,21] further regulating HIF1α- dependent cancer metabolic alterations [22]. In our recently published review, we described a role for the redox activity of APE1 to regulate mitochondrial metabolism, in addition to its role in DNA repair and tumor cell survival and growth. Inhibition of APE1 endonuclease activity is reported to impair mitochondrial respiration in a p53- dependent manner [23]. APE1 when methylated at R301 (Arginine 301) [24] translocates to the mitochondria [25] through the mitochondrial intermembrane space import and assembly protein 40 (MIA) pathway [26] and is responsible for maintaining mitochondrial DNA (mtDNA) integrity under oxidative stress and drives mitochondrial respiration [25]. Other studies have shown that mitochondrial APE1 decreases reactive oxygen species (ROS) generation in osteosarcoma cancer cells thereby promoting cisplatin resistance [27]. Endogenous ROS is a constant threat to mtDNA and reports show that both truncated and full-length APE1 have been found to be capable of repairing mtDNA through its endonuclease activity [28-30].
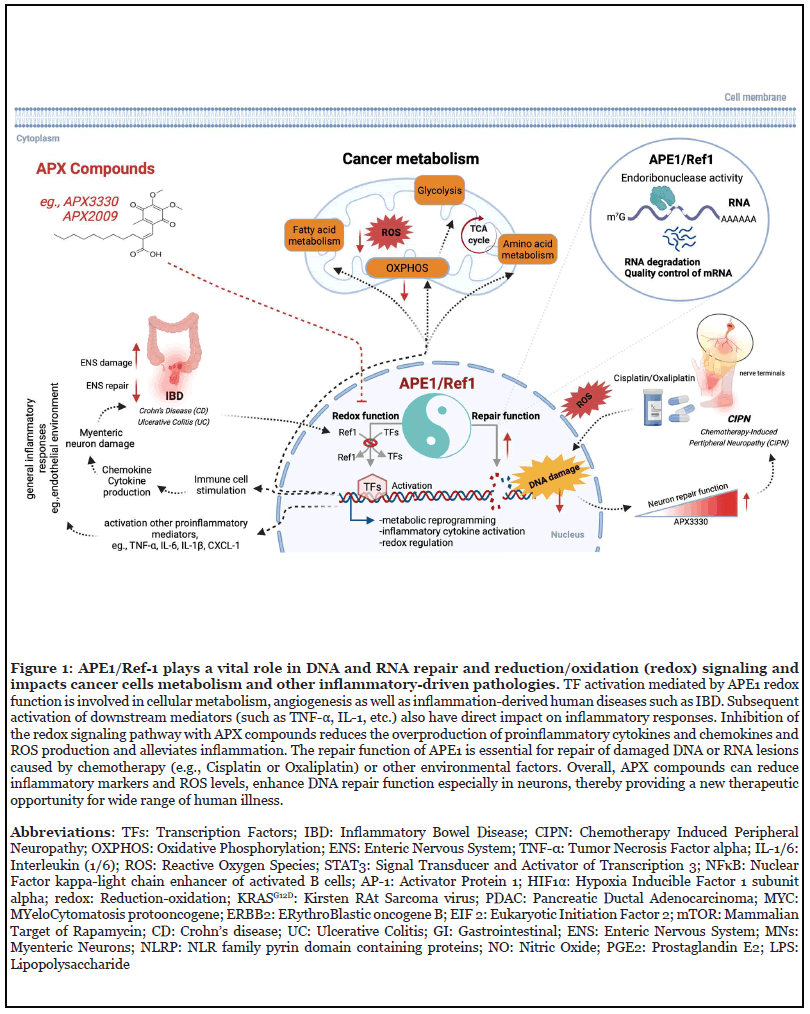
Previously published single cell (sc) RNA-seq analysis of low passage pancreatic cancer patient derived cells (Pa03C) transfected with scrambled or siAPE-1 (small interfering RNA against APE1) revealed novel pathways downregulated with APE1 knockdown including EIF2 as well as mTOR signaling [19]. Under hypoxia, a cluster within scrambled control cells was found to have high expression of HIF1α-regulated genes, whereas that upregulation was abrogated by siAPE1 knockdown. Analysis of the same data identified glycolysis, Tricarboxylic acid (TCA) cycle, and OXPHOS pathways to be downregulated with APE1 knockdown especially under hypoxic conditions (Figure 1) [5].
At the time of publication of the review [1] all of our compiled data as well as the existing literature pointed toward APE1’s repair function within the mitochondria to be predominantly responsible for the observed changes in mitochondrial function. However, more recent data implicate APE1’s redox regulation of transcription factors including HIF1α using APX2009 (a potent second generation APE1 redox inhibitor derived from APX3330) to significantly affect mitochondrial metabolism. Subsequent results from our work revealed that APE1 redox activity strongly influences OXPHOS signaling [6].
All of these findings raise the question: how and what exactly is APE1 orchestrating to control mitochondrial metabolism that regulates and helps provide energy for cancer growth and metastasis? Do these findings suggest that APE1 redox inhibitors alone or in combination with other metabolism altering drugs could provide better treatment options in cancer? This is an area of intense study in our laboratories focusing on the underlying mechanisms of APE1 redox regulation of cancer cells and their mitochondrial metabolic plasticity.
In the Caston et al. review, we discussed the development of the APE1 redox inhibitor APX3330 and second-generation inhibitors APX2009 and APX2014 and others [1]. These compounds specifically inhibit the redox function of APE1; they do not inhibit the DNA repair activity of APE1. In fact, our data suggest that the compounds enhance the endonuclease repair activity of APE1 in sensory and enteric neurons [31,32]. Several publications support the notion that APX3330 and APX2009 are neuroprotective in preclinical disease models and that APE1 plays a role in not just in the tumor cells, but also in CIPN, a common side effect of cancer treatment [31,33-35].
APE1 and Chemotherapy-Induced Peripheral Neuropathy (CIPN)
CIPN is a potentially debilitating side effect of a number of chemotherapy drugs and can lead to a reduction in the dose of chemotherapy thereby potentially limiting efficacy. This neuropathy is characterized by alterations in peripheral sensory function originating in the hands and feet, such as numbness and tingling, increased sensitivity to mechanical touch and cold, loss of proprioception, and reduced tendon reflexes depending on the involvement of sensory and motor nerves. Pain is a debilitating symptom in a subset of patients, who can experience stabbing, burning, and electrical sensations [36]. Treatment and preventative or therapeutic options are limited [37-39].
Previous work strongly supports the hypothesis that DNA damage in neurons is a major contributor to CIPN, especially for platinum-containing drugs (cisplatin, oxaliplatin, and carboplatin). These drugs induce platinum:DNA crosslinks and this adduct formation has been correlated with neuronal toxicity [40,41]. In addition to adduct formation, several platinum drugs also increase the formation of reactive oxygen species (ROS), which leads to oxidative DNA damage and neurotoxicity [19,42-47]. The repair of both endogenous and exogenous DNA damage is critical to maintain neuronal homeostasis [48-51]. Neurons contain a host of DNA repair pathways, including nucleotide excision repair (NER), which is responsible for removing bulky adducts, such as the platinum:DNA crosslinks and the BER pathway, which removes nonbulky damage such as base oxidation and alkylation within nuclear and mitochondrial DNA. Because neurons are highly metabolic [52], there is a steady production of ROS and subsequent oxidative DNA damage, thus the BER pathway predominates as the primary DNA repair pathway in neurons [50,53,54]. APE1 is a key player in BER, hydrolyzing the phosphodiester backbone immediately 5′ to an apurinic/apyrimidinic (AP) site, generating a normal 3′-hydroxyl group and an abasic deoxyribose-5- phosphate, which is processed by subsequent enzymes of the BER pathway. As mentioned above, APE1 is targeted to the mitochondria and is responsible for the repair of both nuclear and mitochondrial DNA. Compromising the repair activity of APE1 would allow oxidative DNA damage to accumulate in neurons, whereas enhancing APE1 repair activity could reduce damage [31,33,34,50].
Our previously published work has demonstrated that overexpression of APE1 protects sensory neurons from oxidative DNA damage, whereas reduction of APE1 expression leads to increased cytotoxicity after neurons were exposed to oxidative DNA damaging agents or chemotherapeutic agents that are known to generate ROS and subsequent oxidative DNA damage [31,33,34,50]. The neuronal protection provided by overexpressing APE1 is recapitulated by APX3330 and APX2009 [33,55-57]. In rat dorsal hind paw skin, sensory nerve-induced vasodilation can be used to assess the function of sensory neurons that innervate the dermis. Systemic administration of cisplatin decreases acute vasodilation induced by sensory nerve activation, whereas treatment of the rats with APX3330 partially restored the vasodilation, suggesting that APX3330 was neuroprotective [31]. Furthermore, the APX compounds do not decrease the efficacy of chemotherapeutics in cancer cell lines or in tumorbearing mice [56]. The ability of the small molecule APX compounds to enhance APE1-mediated DNA repair activity to diminish the neurotoxic effects of chemotherapeutics without compromising antitumor effects provides a novel means to prevent or reverse CIPN [31,34,35].
APE1 in Inflammatory Bowel Disease (IBD)
IBD is characterized by pathological epithelial injury in two main types of intestinal inflammatory conditions: Crohn’s disease (CD) and ulcerative colitis (UC) [58]. In 2015, approximately 1.3% of US adults (3 million) reported being diagnosed with either CD or UC [59]. The UC inflammation is usually induced in the mucosal layer, and typically characterized by chronic bloody diarrhea, tenesmus, and abdominal pain. However, CD describes transmural (full-thickness) inflammation, affecting all layers of the gut wall, can be extended to any part of the gastrointestinal tract (GI) mostly terminal ileum and colon [60]. A long-term complication of IBD is the development of colorectal cancer [58].
GI tract immune dysregulation is a key factor of IBD onset as it is accompanied by a considerable infiltration of inflammatory cells in the gut mucosa [61]. Activation of immune cells and the release of ROS are prominent early events in the pathogenesis of IBD that could dramatically affect the Enteric Nervous System (ENS) [62]. Enteric glial cells are known to actively be involved in inflammatory processes as they can produce and respond to proinflammatory cytokines (e.g., IL-1β, TNF-α, IL-6, etc.) [63]. Myenteric neurons (MNs) are main constituents of ENS located between the longitudinal and circular layers of muscularis externa in the gastrointestinal tract. MNs are also known as major coat of the intestinal wall, mainly responsible for functioning bowel movement. Inflammation and oxidative stress have profound impact on myenteric plexus that could also induce neuronal loss in the colon [64]. For example, in UC rat model, the numbers of MNs were reduced nearly by 50% in comparison to control group [65]. Similar observation was found in a clinial study, there was a significant decrease in the number of MNs (from 604.89 ± 29.65/mm2 to 237.56 ± 59.93/mm2) in patients with UC [66], highlighting the fact that neurotoxic insult on myenteric plexus is a principal cause of IBD. Therefore, the DNA repair and removal mechanisms that cope with oxidative stress-mediated DNA damage must also be considered in addition to the proinflammatory cytokine profile when treating IBD.
Several preclinical investigations demonstrate that APE1 is an effective target in treating IBD [32,67]. For instance, Chang et al. induced IBD in rats with dextran sulfate sodium (DSS) administration, which initially induces colonic injury, and then further causes chronic colitis over the time. They revealed that APE1 and MSH2 (mismatch repair gene) levels were significantly increased in a timedependent manner with DSS-treatment [68]. In addition, DNA damage marker 8-hydroxy-deoxyguanosine (8- OHdG) was increased in the colonic mucosa, whereas APE1 levels in the surface epithelium increased at an earlier timepoint, highlighting the fact that APE1 is a sensitive target for determining exacerbation of DNA damage in DSS-induced colitis [68]. Thus, APE1 may control or contribute to earlier cellular/molecular events in pathogenesis of DSS-induced colitis.
More recent studies by Sahakian et al. established a colitis model in mice that develop defective epithelium which resembles the pathophysiology of human IBD [69]. They demonstrated increased levels of APE1 in both the mucosa and myenteric ganglia as well as overproduction of ROS in mitochondria that caused oxidative DNA damage and translocation of the DNA damage marker HMGB1 from nuclei to cytoplasm in the mouse colons [69]. Furthermore, targeting APE1 redox activity with APX3330 lessened disease severity, reduced immune cell infiltration, restored GI function, and thereby provided neuroprotective effects to the ENS [69]. Most importantly, the repair activity of APE1 was enhanced while APX3330 inhibited the redox activity, suggesting that increases in redox activity and DNA damage both underlie the pathological bowel inflammation and provides a potential therapeutic approach for treating IBD. This finding of APE1 redox inhibition by APX3330 with enhancement of APE1 repair function is similar to what was seen in dorsal root ganglion (DRG) neurons in the CIPN models [31,33-35].
APE1/Ref-1 in Inflammation-Associated Diseases
Inflammation is a biological response of the immune system that is induced by various invading pathogens and/or endogenous stimuli and essential for tissue healing processes [70]. Chronic inflammation, however, disrupts cellular homeostasis and leads to diseases, such as cardiovascular and bowel diseases, diabetes, osteoarthritis, and various cancers [71].
Cytokines are cell signaling proteins produced primarily by immune cells, including macrophages, dendritic cells, lymphocytes, tumors, and cancer associated fibroblasts in response to different stimulation [71,72]. However, certain cytokines, such as tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6), can be also induced by tumor cells as well as non-cancerous cell types, such as epithelial cells or cancer-associated fibroblasts (CAFs) [73]. A “cytokine storm” is an event that describes activation of an aggressive inflammatory response with the release of a large amount of pro-inflammatory cytokines, such as IL- 1, TNF-α, and IL-6 [74]. The release of these cytokines is a major driver in most inflammatory disorders, including Severe Acute Respiratory Syndrome Coronavirus 2 (SARSCoV- 2) (COVID-19) [75]. Increasing numbers of clinical studies support that cytokine dynamics directly correlate to the clinical severity of COVID-19 infections, suggesting a central role of pro-inflammatory cytokines in respiratory diseases and activation of the adaptive immune response [76]. Cytokine receptor activation triggers important intracellular signaling pathways, including the mitogenactivated protein kinase (MAPK), nuclear factor kappa-B (NF-κB), and Janus kinase (JAK)-signal transducer and activator of transcription (STAT). For instance, more than 50 cytokines (such as IL-6 and others) found to be regulated via JAK/STAT signaling cascade induces inflammation and control the immune response [77]. Similarly, several other cytokines (such as TNF-α, IL-1 and IL-17) signaling cascades partially overlap and utilize some of the same mediators leading to the activation of some common TFs, such as NF-κB, AP-1, STAT3, ultimately leading to the regulation of gene expression [78].
As previously mentioned, APE1 is a redox catalyst involved with various biological processes by virtue of reducing several important TFs to enhance their binding to DNA, including STAT3, NF-κB, AP-1 and others involved in inflammatory regulation and response [19,30,79]. APE1 mediation of these key TFs and their activation of cytokines, chemokines, and other downstream targets promote growth, migration, and survival in tumor cells as well as angiogenesis in the tumor microenvironment [80]. A bioinformatics analysis demonstrated potential AP-1 and NF-κB binding sites within the promoter sequences of NLR family pyrin domain containing proteins (NLRP), namely NLRP1 and NLRP3, which are key biomarkers of multiprotein intracellular complexes known as inflammasomes [81]. Indeed, Tang et al. demonstrated that APE1 promoted lipopolysaccharide (LPS)-induced NLRP3 inflammasome activation, along with overproduction of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL- 18 in tumor-associated macrophages through an NFκBdependent pathway. In the same study, they showed that transcriptional levels of TNF-α and NLRP3 along with other inflammasomes-associated molecules Caspase-1 and ASC were significantly inhibited by both APX3330 and APE1 protein knockdown in THP-1 cells, suggesting the involvement of an APE1-NLRP3 axis in inflammasome pathways [82]. Jedinak et al. demonstrated that APX3330 suppressed secretion of inflammatory cytokines, including TNF-α, IL-6, IL-12 and the inflammatory mediator nitric oxide (NO) as well as prostaglandin E2 (PGE2) in LPSstimulated macrophages through inhibition of TFs NF-κB and AP-1, both well-known primary targets of APE1 [83]. Taken together, these studies underscore the regulatory role of APE1 in various inflammatory processes by controlling important TFs that mediate proinflammatory cytokines.
APE1 Role in RNA Metabolism
RNA is susceptible to base oxidation and alkylation, but how this damage is resolved is not fully understood. Some damage may be repaired via the protein hABH3RNA. However, some processing will likely occur through RNA metabolism to remove mutated RNA strands and damaged nucleotides from the RNA pool. Damaged RNA can lead to defective or mutant proteins during translation and likely inhibit reverse transcriptase activity, potentially contributing to cancer, schizophrenia, or muscle dystrophy [84,85].
APE1 has been characterized as a regulator of mRNA, which removes lesions from oxidative RNA substrates [86]. When APE1 is knocked down in HeLa cells, the number of oxidized mRNA increased, implying that APE1 is involved in mRNA cleansing [86]. APE1 is also capable of cleaving mRNA using its endonuclease activity, causing RNA degradation [86-90]. Currently, APE1 is known to cleave cMyc, SARS and CD44 [90].
APE1 also enhances post-translational maturation of microRNA. Specifically, APE1 has been shown to interact with the DROSHA complex to cleave microRNA-221/222, an oncogenic miRNA when upregulated, causing thyroid papillary carcinomas, glioblastomas, prostate carcinoma, and gastric carcinoma [91]. Binding of APE1 to DROSHA was increased under conditions of oxidative stress. Increased cleavage of pre-miRNA-221/222 by APE1 led to cancer cell growth in vitro. A similar result was seen with miR-92b, which enhances cervical carcinoma [92]. Therefore, dysregulation of APE1’s endonuclease function further supports the evidence that APE1 plays a role in not only DNA repair, but RNA metabolism as a cleansing mechanism of damaged RNA, with implications for cancer progression.
Summary and Conclusion
In this review, we have highlighted the recent advances in our understanding of APE1 signaling in various pathological conditions, namely CIPN, IBD, as well as general inflammatory signaling along with possible pharmacological interventions (Figure 1). Initially, we discussed how APE1’s redox function alters mitochondrial metabolism during cancer progression. APE1 is present in both the nucleus and the mitochondria; its impact on metabolism could stem from its role in DNA repair as well as its role in regulating the activity of various TFs involved in gene regulation.
Additionally, we focused on APE1 in neuronal pathologies, we have discussed that selective inhibition of APE1’s redox function, without impeding the repair function, leads to increased DNA repair activity of APE1. In CIPN, the neurotoxic effects of platinating agents were diminished without compromising their anti-tumor properties through inhibition of APE1’s redox signaling. Similarly, in IBD, inhibiting APE1’s redox signaling had a neuroprotective effect, as well as anti-inflammatory, lessening the disease severity and enhancing APE1 DNA repair capacity, specifically with a reduction in oxidative DNA damage.
Finally, we explored APE1’s role in microRNA maturation and therein disease progression. Unlike CIPN and IBD, which have been linked to mis-regulated redox signaling, APE1’s interaction with microRNAs is dependent on its endonuclease activity.
In conclusion, APE1’s redox and DNA repair capabilities are interwoven in the cellular response to cancer metabolism, neuropathy, inflammation, and RNA processing. APE1 is a “hotspot” for inflammatory-oxidative connections, which provide a new therapeutic opportunity for treating a wide range of debilitating human diseases.
Conflict of Interest
MRK is a member of the Ocuphire medical advisory board and CSO and co-founder of Apexian Pharmaceuticals which licensed APX3330 and other APX compounds from Indiana University School of Medicine. Ocuphire Pharma licensed APX3330 for ocular and diabetes indications from Apexian Pharmaceuticals.
Funding
M.R.K. and M.L.F. were supported by grants from the National Institute of Health and National Cancer Institute R01CA167291 and R01CA167291-S1. Dr. Kelley was also supported by NIH/NCI grants R01CA205166, R01CA231267, R01 EY031939 and R01HL140961. Dr. Fishel was supported by NIH/NCI grant U01HL143403. Dr. Fishel was also supported by R01CA211098 and R01NF180045. M.L.F. and M.R.K. were additionally supported by the Riley Children’s Foundation.
Acknowledgement
The figure was created using BioRender (BioRender. com).
Author Contributions
All authors contributed to writing, reviewing, and editing manuscript. MM contributed to figure creation and construction and editing. MRK also contributed to figure creation.
References
2. El Hadri K, Mahmood DF, Couchie D, Jguirim-Souissi I, Genze F, Diderot V, et al. Thioredoxin-1 promotes antiinflammatory macrophages of the M2 phenotype and antagonizes atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32(6):1445-52.
3. Chen B, Guan D, Cui ZJ, Wang X, Shen X. Thioredoxin 1 downregulates MCP-1 secretion in human endothelial cells by suppressing nuclear Translocation of activator protein 1 and redox factor-1. Am J Physiol Cell Physiol. 2010;298(5):C1170-9.
4. Sriramajayam K, Peng D, Lu H, Zhou S, Bhat N, McDonald OG, et al. Activation of NRF2 by APE1/REF1 is redox-dependent in Barrett’s related esophageal adenocarcinoma cells. Redox biology. 2021;43:101970.
5. Gampala S, Shah F, Lu X, Moon H-r, Sandusky GE, Hulsey E, et al., editors. Ref-1 redox function identified as mitochondrial metabolic regulator in pancreatic cancer cells but not in CAFs2021 March 1 2021: Cancer Research.
6. Silpa G, Fenil S, Xiaoyu L, Hye-ran M, George S, Emily H, et al. Ref-1 Redox Activity Alters Cancer Cell Metabolism in Pancreatic Cancer: Exploiting This Novel Finding as a Potential Target. Journal of Experimental & Clinical Cancer Research. 2021.
7. Sahakian L, Filippone RT, Stavely R, Robinson AM, Yan XS, Abalo R, et al. Inhibition of APE1/Ref-1 Redox Signaling Alleviates Intestinal Dysfunction and Damage to Myenteric Neurons in a Mouse Model of Spontaneous Chronic Colitis. Inflammatory Bowel Diseases. 2020;27(3):388-406.
8. Curtis Heisel JY, Mahmut Mijiti, Kostas Charizanis, Mitchel Brigell, Timothy W. Corson, Mark R. Kelley APE1/ Ref-1 as a Novel Target for Retinal Diseases. Journal of Cell Signaling. 2021;2(2):133-8.
9. Sardar Pasha SPB, Sishtla K, Sulaiman RS, Park B, Shetty T, Shah F, et al. Ref-1/APE1 Inhibition with Novel Small Molecules Blocks Ocular Neovascularization. J Pharmacol Exp Ther. 2018;367(1):108-18.
10. Tell G, Damante G, Caldwell D, Kelley MR. The intracellular localization of APE1/Ref-1: more than a passive phenomenon? Antioxid Redox Signal. 2005;7(3- 4):367-84.
11. Di Maso V, Avellini C, Crocè LS, Rosso N, Quadrifoglio F, Cesaratto L, et al. Subcellular Localization of APE1/Ref-1 in Human Hepatocellular Carcinoma: Possible Prognostic Significance. Molecular Medicine. 2007;13(1-2):89-96.
12. Nath S, Roychoudhury S, Kling M, Song H, Biswas P, Shukla A, et al. The extracellular role of DNA damage repair protein APE1 in regulation of IL-6 expression. Cellular Signalling. 2017.
13. Shin JH, Choi S, Lee YR, Park MS, Na YG, Irani K, et al. APE1/Ref-1 as a Serological Biomarker for the Detection of Bladder Cancer. Cancer Research and Treatment : official journal of Korean Cancer Association. 2015;47(4):823-33.
14. Choi S, Shin JH, Lee YR, Joo HK, Song KH, Na YG, et al. Urinary APE1/Ref-1: A Potential Bladder Cancer Biomarker. Disease Markers. 2016;2016:7276502.
15. Pascut D, Sukowati C, Antoniali G, Mangiapane G, Burra S, Giovanni Mascaretti L, et al. Serum AP-endonuclease 1 (sAPE1) as novel biomarker for hepatocellular carcinoma. Oncotarget. 2019 Jan 8;10(3):383-394.
16. Lee YR, Joo HK, Jeon BH. The Biological Role of Apurinic/Apyrimidinic Endonuclease1/Redox Factor-1 as a Therapeutic Target for Vascular Inflammation and as a Serologic Biomarker. Biomedicines. 2020;8(3).
17. Porporato PE, Filigheddu N, Pedro JMB-S, Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer. Cell research. 2018;28(3):265-80.
18. Alameddine AK, Conlin FT, Binnall BJ, Alameddine YA, Alameddine KO. How do cancer cells replenish their fuel supply? Cancer Reports. 2018;1(1):e1003.
19. Shah F, Logsdon D, Messmann RA, Fehrenbacher JC, Fishel ML, Kelley MR. Exploiting the Ref-1-APE1 node in cancer signaling and other diseases: from bench to clinic. NPJ Precision Oncology. 2017;1:19.
20. Logsdon DP, Shah F, Carta F, Supuran CT, Kamocka M, Jacobsen MH, et al. Blocking HIF signaling via novel inhibitors of CA9 and APE1/Ref-1 dramatically affects pancreatic cancer cell survival. Sci Rep. 2018;8(1):13759- 73.
21. Logsdon DP GM, Luo M, Shahda S, Jiang Y, Tong Y, Yu Z, Zyromski N, et al. Regulation of HIF1a under Hypoxia by APE1/Ref-1 Impacts CA9 Expression: Dual-Targeting in Patient-Derived 3D Pancreatic Cancer Models. Molecular Cancer Therapeutics. 2016;15(11):2722-32.
22. Zhang C, Ward J, Dauch JR, Tanzi RE, Cheng HT. Cytokine-mediated inflammation mediates painful neuropathy from metabolic syndrome. Plos One. 2018;13(2):e0192333.
23. Codrich M, Comelli M, Malfatti MC, Mio C, Ayyildiz D, Zhang C, et al. Inhibition of APE1-endonuclease activity affects cell metabolism in colon cancer cells via a p53- dependent pathway. DNA Repair. 2019;82:102675.
24. Zhang Y, Zhang Q, Li L, Mu D, Hua K, Ci S, et al. Arginine methylation of APE1 promotes its mitochondrial translocation to protect cells from oxidative damage. Free Radical Biology and Medicine. 2020.
25. Barchiesi A, Bazzani V, Tolotto V, Elancheliyan P, Wasilewski M, Chacinska A, et al. Mitochondrial oxidative stress induces rapid intermembrane space/matrix translocation of Apurinic/apyrimidinic endonuclease 1 protein through TIM23 complex. Journal of Molecular Biology. 2020.
26. Barchiesi A, Wasilewski M, Chacinska A, Tell G, Vascotto C. Mitochondrial translocation of APE1 relies on the MIA pathway. Nucleic Acids Res. 2015;43(11):5451-64.
27. Liu Y, Zhang Z, Zhang L, Zhong Z. Cytoplasmic APE1 promotes resistance response in osteosarcoma patients with cisplatin treatment. Cell biochemistry and function.2020;38(2):195-203.
28. Chattopadhyay R, Wiederhold L, Szczesny B, Boldogh I, Hazra TK, Izumi T, et al. Identification and characterization of mitochondrial abasic (AP)- endonuclease in mammalian cells. Nucleic Acids Res. 2006;34(7):2067-76.
29. Li M-X, Wang D, Zhong Z-Y, Xiang D-B, Li Z-P, Xie J-Y, et al. Targeting truncated APE1 in mitochondria enhances cell survival after oxidative stress. Free Radical Biology and Medicine. 2008;45(5):592-601.
30. Tell G, Crivellato E, Pines A, Paron II, Pucillo C, Manzini G, et al. Mitochondrial localization of APE/Ref-1 in thyroid cells. Mutat Res. 2001;485(2):143-52.
31. Kelley MR, Jiang YL, Guo CL, Reed A, Meng HD, Vasko MR. Role of the DNA Base Excision Repair Protein, APE1 in Cisplatin, Oxaliplatin, or Carboplatin Induced Sensory Neuropathy. Plos One. 2014;9(9).
32. Sahakian L, Filippone RT, Stavely R, Robinson AM, Yan XS, Abalo R, et al. Inhibition of APE1/Ref-1 redox signalling alleviates intestinal dysfunction and damage to myenteric neurons in a mouse model of spontaneous chronic colitis. Inflammatory Bowel Diseases2020.
33. Fehrenbacher JC, Guo C, Kelley MR, Vasko MR. DNA damage mediates changes in neuronal sensitivity induced by the inflammatory mediators, MCP-1 and LPS, and can be reversed by enhancing the DNA repair function of APE1. Neuroscience. 2017;366:23-35.
34. Kelley MR, Wikel JH, Guo C, Pollok KE, Bailey BJ, Wireman R, et al. Identification and Characterization of New Chemical Entities Targeting Apurinic/Apyrimidinic Endonuclease 1 for the Prevention of Chemotherapy- Induced Peripheral Neuropathy. J Pharmacol Exp Ther. 2016;359(2):300-9.
35. Kim HS, Guo C, Thompson EL, Jiang Y, Kelley MR, Vasko MR, et al. APE1, the DNA base excision repair protein, regulates the removal of platinum adducts in sensory neuronal cultures by NER. Mutat Res. 2015;779:96-104.
36. Seretny M, Currie GL, Sena ES, Ramnarine S, Grant R, MacLeod MR, et al. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: A systematic review and meta-analysis. Pain. 2014;155(12):2461-70.
37. Hershman DL, Lacchetti C, Dworkin RH, Lavoie Smith EM, Bleeker J, Cavaletti G, et al. Prevention and Management of Chemotherapy-Induced Peripheral Neuropathy in Survivors of Adult Cancers: American Society of Clinical Oncology Clinical Practice Guideline. Journal of Clinical Oncology. 2014;32(18):1941-67.
38. Stone JB, DeAngelis LM. Cancer-treatment-induced neurotoxicity[mdash]focus on newer treatments. Nat Rev Clin Oncol. 2016;13(2):92-105.
39. Hu S, Huang KM, Adams EJ, Loprinzi CL, Lustberg MB. Recent Developments of Novel Pharmacologic Therapeutics for Prevention of Chemotherapy-Induced Peripheral Neuropathy. Clin Cancer Res. 2019.
40. Ta LE, Espeset L, Podratz J, Windebank AJ. Neurotoxicity of oxaliplatin and cisplatin for dorsal root ganglion neurons correlates with platinum-DNA binding. NeuroToxicology. 2006;27(6):992-1002.
41. Dzagnidze A, Katsarava Z, Makhalova J, Liedert B, Yoon MS, Kaube H, et al. Repair capacity for platinum- DNA adducts determines the severity of cisplatin-induced peripheral neuropathy. J Neurosci. 2007;27(35):9451-7.
42. Kelley MR, Fehrenbacher JC. Challenges and opportunities identifying therapeutic targets for chemotherapy-induced peripheral neuropathy resulting from oxidative DNA damage. Neural Regen Res. 2017;12(1):72-4.
43. Kleih M, Böpple K, Dong M, Gaißler A, Heine S, Olayioye MA, et al. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell death & disease. 2019;10(11):1-12.
44. Cruz-Bermúdez A, Laza-Briviesca R, Vicente-Blanco RJ, García-Grande A, Coronado MJ, Laine-Menéndez S, et al. Cisplatin resistance involves a metabolic reprogramming through ROS and PGC-1a in NSCLC which can be overcome by OXPHOS inhibition. Free Radical Biology and Medicine. 2019;135:167-81.
45. Xue D-F, Pan S-T, Huang G, Qiu J-X. ROS enhances the cytotoxicity of cisplatin by inducing apoptosis and autophagy in tongue squamous cell carcinoma cells. The International Journal of Biochemistry & Cell Biology. 2020;122:105732.
46. Siomek A, Tujakowski J, Gackowski D, Rozalski R, Foksinski M, Dziaman T, et al. Severe oxidatively damaged DNA after cisplatin treatment of cancer patients. Int J Cancer. 2006.
47. Preston TJ, Henderson JT, McCallum GP, Wells PG. Base excision repair of reactive oxygen speciesinitiated 7,8-dihydro-8-oxo-2'-deoxyguanosine inhibits the cytotoxicity of platinum anticancer drugs. Mol Cancer Ther. 2009;8(7):2015-26.
48. Brooks PJ. DNA repair in neural cells: basic science and clinical implications. Mutat Res. 2002;509(1-2):93- 108.
49. McMurray CT. To die or not to die: DNA repair in neurons. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2005;577(1-2):260-74.
50. Fishel ML, Vasko MR, Kelley MR. DNA repair in neurons: so if they don't divide what's to repair? Mutat Res. 2007;614(1-2):24-36.
51. Hetman M, Vashishta A, Rempala G. Neurotoxic mechanisms of DNA damage: focus on transcriptional inhibition. J Neurochem. 2010;114(6):1537-49.
52. Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997;77(3):731-58.
53. Barzilai A, Biton S, Shiloh Y. The role of the DNA damage response in neuronal development, organization and maintenance. DNA Repair (Amst). 2008;7(7):1010-27.
54. Fortini P, Dogliotti E. Mechanisms of dealing with DNA damage in terminally differentiated cells. Mutat Res.2010;685(1-2):38-44.
55. Vasko MR, Guo C, Thompson EL, Kelley MR. The repair function of the multifunctional DNA repair/redox protein APE1 is neuroprotective after ionizing radiation. DNA Repair (Amst). 2011;10(9):942-52.
56. Kelley MR, Jiang Y, Guo C, Reed A, Meng H, Vasko MR. Role of the DNA base excision repair protein, APE1 in cisplatin, oxaliplatin, or carboplatin induced sensory neuropathy. Plos One. 2014;9(9):e106485.
57. Kelley MR, Wikel JH, Guo C, Pollok KE, Bailey BJ, Wireman R, et al. Identification and Characterization of new chemical entities targeting Apurinic/Apyrimidinic Endonuclease 1 for the prevention of chemotherapyinduced peripheral neuropathy (CIPN). J Pharmacol Exp Ther. 2016;359(2):300-9.
58. Rubin DC, Shaker A, Levin MS. Chronic intestinal inflammation: inflammatory bowel disease and colitisassociated colon cancer. Front Immunol. 2012;3:107.
59. Dahlhamer JM, Zammitti EP, Ward BW, Wheaton AG, Croft JB. Prevalence of Inflammatory Bowel Disease Among Adults Aged >/=18 Years - United States, 2015. MMWR Morb Mortal Wkly Rep. 2016;65(42):1166-9.
60. Ashktorab H, Brim H, Hassan S, Nouraie M, Gebreselassie A, Laiyemo AO, et al. Inflammatory polyps occur more frequently in inflammatory bowel disease than other colitis patients. BMC Gastroenterol. 2020;20(1):170.
61. Ahluwalia B, Moraes L, Magnusson MK, Ohman L. Immunopathogenesis of inflammatory bowel disease and mechanisms of biological therapies. Scand J Gastroenterol. 2018;53(4):379-89.
62. Lakhan SE, Kirchgessner A. Neuroinflammation in inflammatory bowel disease. J Neuroinflammation.2010;7:37.
63. Ochoa-Cortes F, Turco F, Linan-Rico A, Soghomonyan S, Whitaker E, Wehner S, et al. Enteric Glial Cells: A New Frontier in Neurogastroenterology and Clinical Target for Inflammatory Bowel Diseases. Inflamm Bowel Dis. 2016;22(2):433-49.
64. Shang B, Shi H, Wang X, Guo X, Wang N, Wang Y, et al. Protective effect of melatonin on myenteric neuron damage in experimental colitis in rats. Fundam Clin Pharmacol. 2016;30(2):117-27.
65. Sanovic S, Lamb DP, Blennerhassett MG. Damage to the Enteric Nervous System in Experimental Colitis. The American Journal of Pathology. 1999;155(4):1051-7.
66. Bernardini N, Segnani C, Ippolito C, De Giorgio R, Colucci R, Faussone-Pellegrini MS, et al. Immunohistochemical analysis of myenteric ganglia and interstitial cells of Cajal in ulcerative colitis. J Cell Mol Med. 2012;16(2):318-27.
67. Chang IY, Kim JN, Maeng YH, Yoon SP. Apurinic/ apyrimidinic endonuclease 1, the sensitive marker for DNA deterioration in dextran sulfate sodium-induced acute colitis. Redox Rep. 2013.
68. Chang IY, Kim JN, Maeng YH, Yoon SP. Apurinic/ apyrimidinic endonuclease 1, the sensitive marker for DNA deterioration in dextran sulfate sodium-induced acute colitis. Redox Rep. 2013;18(5):165-73.
69. Sahakian L, Filippone RT, Stavely R, Robinson AM, Yan XS, Abalo R, et al. Inhibition of APE1/Ref-1 Redox Signaling Alleviates Intestinal Dysfunction and Damage to Myenteric Neurons in a Mouse Model of Spontaneous Chronic Colitis. Inflamm Bowel Dis. 2021;27(3):388-406.
70. Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 2018;9(6):7204-18.
71. Varade J, Magadan S, Gonzalez-Fernandez A. Human immunology and immunotherapy: main achievements and challenges. Cell Mol Immunol. 2021;18(4):805-28.
72. Ray A. Cytokines and their Role in Health and Disease: A Brief Overview. MOJ Immunology. 2016;4(2).
73. Burkholder B, Huang RY, Burgess R, Luo S, Jones VS, Zhang W, et al. Tumor-induced perturbations of cytokines and immune cell networks. Biochim Biophys Acta. 2014;1845(2):182-201.
74. Peter AE, Sandeep BV, Rao BG, Kalpana VL. Calming the Storm: Natural Immunosuppressants as Adjuvants to Target the Cytokine Storm in COVID-19. Front Pharmacol. 2020;11:583777.
75. Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20(6):355-62.
76. Ragab D, Salah Eldin H, Taeimah M, Khattab R, Salem R. The COVID-19 Cytokine Storm; What We Know So Far. Front Immunol. 2020;11:1446.
77. Morris R, Kershaw NJ, Babon JJ. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018;27(12):1984-2009.
78. Gadina M, Gazaniga N, Vian L, Furumoto Y. Small molecules to the rescue: Inhibition of cytokine signaling in immune-mediated diseases. J Autoimmun. 2017;85:20-31.
79. Cai Z, Kotzin JJ, Ramdas B, Chen S, Nelanuthala S, Palam LR, et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell stem cell. 2018;23(6):833-49.e5.
80. Jiang X, Wang J, Deng X, Xiong F, Zhang S, Gong Z, et al. The role of microenvironment in tumor angiogenesis. J Exp Clin Cancer Res. 2020;39(1):204.
81. Maharana J, Panda D, De S. Deciphering the ATPbinding mechanism(s) in NLRP-NACHT 3D models using structural bioinformatics approaches. Plos One.2018;13(12):e0209420.
82. Tang Z, Wang Y, Wan Y, Xie Y, Li S, Tao D, et al. Apurinic/apyrimidinic endonuclease 1/reductionoxidation effector factor-1 (APE1) regulates the expression of NLR family pyrin domain containing 3 (NLRP3) inflammasome through modulating transcription factor NF-kappaB and promoting the secretion of inflammatory mediators in macrophages. Ann Transl Med. 2021;9(2):145.
83. Jedinak A, Dudhgaonkar S, Kelley MR, Sliva D. Apurinic/Apyrimidinic endonuclease 1 regulates inflammatory response in macrophages. Anticancer Res. 2011;31(2):379-85.
84. Wurtmann EJ, Wolin SL. RNA under attack: cellular handling of RNA damage. Crit Rev Biochem Mol Biol. 2009;44(1):34-49.
85. Aznaourova M, Schmerer N, Schmeck B, Schulte LN. Disease-Causing Mutations and Rearrangements in Long Non-coding RNA Gene Loci. Frontiers in Genetics. 2020;11:527484.
86. Antoniali G, Serra F, Lirussi L, Tanaka M, D’Ambrosio C, Zhang S, et al. Mammalian APE1 controls miRNA processing and its interactome is linked to cancer RNA metabolism. Nature Communications. 2017;8(1):797-815.
87. Chohan M, Mackedenski S, Li WM, Lee CH. Human apurinic/apyrimidinic endonuclease 1 (APE1) has 3’ RNA phosphatase and 3’ exoribonuclease activities. J Mol Biol. 2015;427(2):298-311.
88. Kim WC, King D, Lee CH. RNA-cleaving properties of human apurinic/apyrimidinic endonuclease 1 (APE1). Int J Biochem Mol Biol. 2010;1(1):12-25.
89. Jobert L, Nilsen H. Regulatory mechanisms of RNA function: emerging roles of DNA repair enzymes. Cell Mol Life Sci. 2014;71(13):2451-65.
90. Kim W-C, King D, Lee CH. RNA-cleaving properties of human apurinic/apyrimidinic endonuclease 1 (APE1). Int J Biochem Mol Biol. 2010;1(1):12-25.
91. Garofalo M, Quintavalle C, Romano G, Croce CM, Condorelli G. miR221/222 in Cancer: Their Role in Tumor Progression and Response to Therapy. Current Molecular Medicine. 2012;12(1):27-33.
92. Sun Y, Feng Y, Zhang G, Xu Y. The endonuclease APE1 processes miR-92b formation, thereby regulating expression of the tumor suppressor LDLR in cervical cancer cells. Ther Adv Med Oncol. 2019;11:1758835919855859.