Abstract
Current therapies for antibody-mediated autoimmune diseases largely rely on broad immunosuppression or lineage-wide B-cell depletion. These approaches are associated with increased risks of infection and other adverse effects. This commentary focuses on Chimeric Autoantibody Receptor (CAAR) T-cell technology as a precision immunotherapy that selectively eliminates pathogenic autoreactive B-cell clones while preserving protective immunity. Using Pemphigus Vulgaris (PV) as a representative model, CAAR-T cells engineered to express desmoglein 3 (Dsg3) demonstrate antigen-specific targeting of B cells expressing anti-Dsg3 B cell receptors. Notably, these cells maintain cytolytic efficacy despite high circulating autoantibody titers and show tissue selectivity, sparing keratinocytes, potentially due to biophysical constraints governing immune synapse formation. We further discuss key translational challenges, including manufacturing complexity, the presence of additional pathogenic antibody populations (e.g., anti-Dsg1), and long-term cellular persistence. Collectively, CAAR-T technology represents a mechanistically distinct and potentially scalable therapeutic paradigm for antibody-mediated autoimmune disorders, emphasizing antigen-specific immune editing over global immune suppression.
Keywords
Pemphigus, Dsg3, CAAR T-cell therapy, Autoimmune diseases
Introduction
Treating autoimmune diseases presents a significant therapeutic challenge, as pathogenic immune responses are often intertwined with protective immunity required to protect against cancer and infections [1,2]. Most therapies currently available rely on broad immunosuppression or whole immune cell depletion, which effectively reduce disease activity but introduce significant risks, including infection, malignancy, and long-term immune dysregulation [3,4]. The development of approaches that selectively eliminate autoreactive immune cells, particularly autoreactive B cells, without compromising systemic immune competence, has emerged as a central objective in translational immunology [5].
Recently, a landmark study by Ellebrecht and colleagues, published in Science, introduced an elegant solution to this long-standing problem by engineering chimeric autoantibody receptor (CAAR) T cells [6]. Using Pemphigus Vulgaris (PV) as a model antibody-mediated autoimmune disease, their work demonstrated that antigen-based targeting could selectively recognize and eliminate pathogenic B cells. Rather than relying on generalized immune suppression, this approach reframes autoimmune therapy as antigen-specific immune editing.
Pemphigus Vulgaris as a Precision Targeting Model
Pemphigus Vulgaris (PV) is a prototypic antibody-mediated autoimmune disease driven by IgG autoantibodies directed against desmoglein 3 (Dsg3), a cadherin adhesion molecule essential for keratinocyte cohesion [7,8]. Pathogenic autoantibodies disrupt desmosomal integrity, leading to intraepidermal blistering [9]. Importantly, the pathogenic role of anti-Dsg3 antibodies has been supported by a series of studies, such as passive transfer models, serological correlations, and therapeutic responses [10–12].
Clinical success with CD20-directed B-cell depletion, notably with rituximab, further confirmed the central role of B cells in pemphigus pathogenesis [13]. However, relapse remains frequent, largely due to persistence or reconstitution of autoreactive memory B-cell clones [14]. Moreover, global B-cell depletion compromises protective humoral immunity [13,15], highlighting the need for selective and more effective targeting strategies.
PV represents a uniquely suitable model for antigen-specific immunotherapy for several reasons. First, the disease is defined by well-characterized autoantigens (Dsg1 and Dsg3) [8,10], enabling precise molecular targeting. Second, pathogenicity is directly dependent on the B-cell receptor (BCR) and circulating autoantibody specificity [11,12], linking disease mechanisms to targetable cellular populations. Third, autoreactive B-cell clones can be detected and functionally characterized [16]. Finally, the mechanisms underlying clinical relapse are increasingly understood [17], providing a mechanistic framework for evaluating targeted interventions.
Conceptual Innovation of CAAR-T cells
Traditional CAR-T cell therapy, originally developed for oncology, targets lineage-restricted surface markers such as CD19 using a single-chain variable fragment (ScFv) of specific antibodies [18–20] (Table 1). This works well in malignancy, where eliminating an entire cell lineage is acceptable. In contrast, autoimmune diseases are characterized by specificity rather than lineage. CAR19 T cells have been shown to achieve disease remission by enhancing and sustaining B cell depletion in preclinical and clinical trials across various autoimmune diseases [21–23]. Although a series of clinical trials focused on these global B-cell lineages has been underway recently (Figure 1), CAAR-T cells fundamentally invert this targeting paradigm [6]. Rather than recognizing a conventional surface antigen, the engineered receptor incorporates the autoantigen itself. In the context of PV, Dsg3 functions as the extracellular recognition domain, enabling T cells to selectively engage B cells expressing anti-Dsg3 BCRs [6,16].
This design introduces several important advantages. Most notably, cellular specificity is defined by antigen recognition rather than phenotype, thereby allowing selective identification of pathogenic clones. This design enables targeted killing of autoreactive B cells while preserving the broader protective B-cell repertoire. Furthermore, CAAR-T therapy is a form of functional immune editing that reshapes autoreactive compartments rather than globally suppressing immune responses.
|
Feature |
CAR T Therapy |
CAAR T Therapy |
|
T-cell Type |
Chimeric Antigen Receptor T-cell (CAR T-cell) |
Chimeric Autoantibody Receptor T-cell (CAAR T-cell) |
|
Primary Target |
Cells expressing a specific antigen (e.g., CD19 on B-cell lymphoma). |
B-cells expressing a specific surface antibody (BCR) that attacks the body. |
|
Extracellular Domain |
Usually a single-chain variable fragment (scFv) derived from an antibody. |
The specific antigen that the rogue autoantibody usually attacks. |
|
Mechanism of Action |
Recognizes and kills all cells carrying the target antigen (often including healthy cells). |
Acts as "bait" to bind only to the specific B-cells producing autoantibodies. |
|
Main Clinical Use |
Hematologic cancers (Leukemia, Lymphoma, Myeloma). |
Autoimmune diseases (e.g., Pemphigus Vulgaris, Myasthenia Gravis). |
|
Off-Target Effects |
B-cell aplasia: Kills all B-cells (both healthy and cancerous). |
Targeted: Ideally leaves healthy B-cells intact, preserving general immunity. |
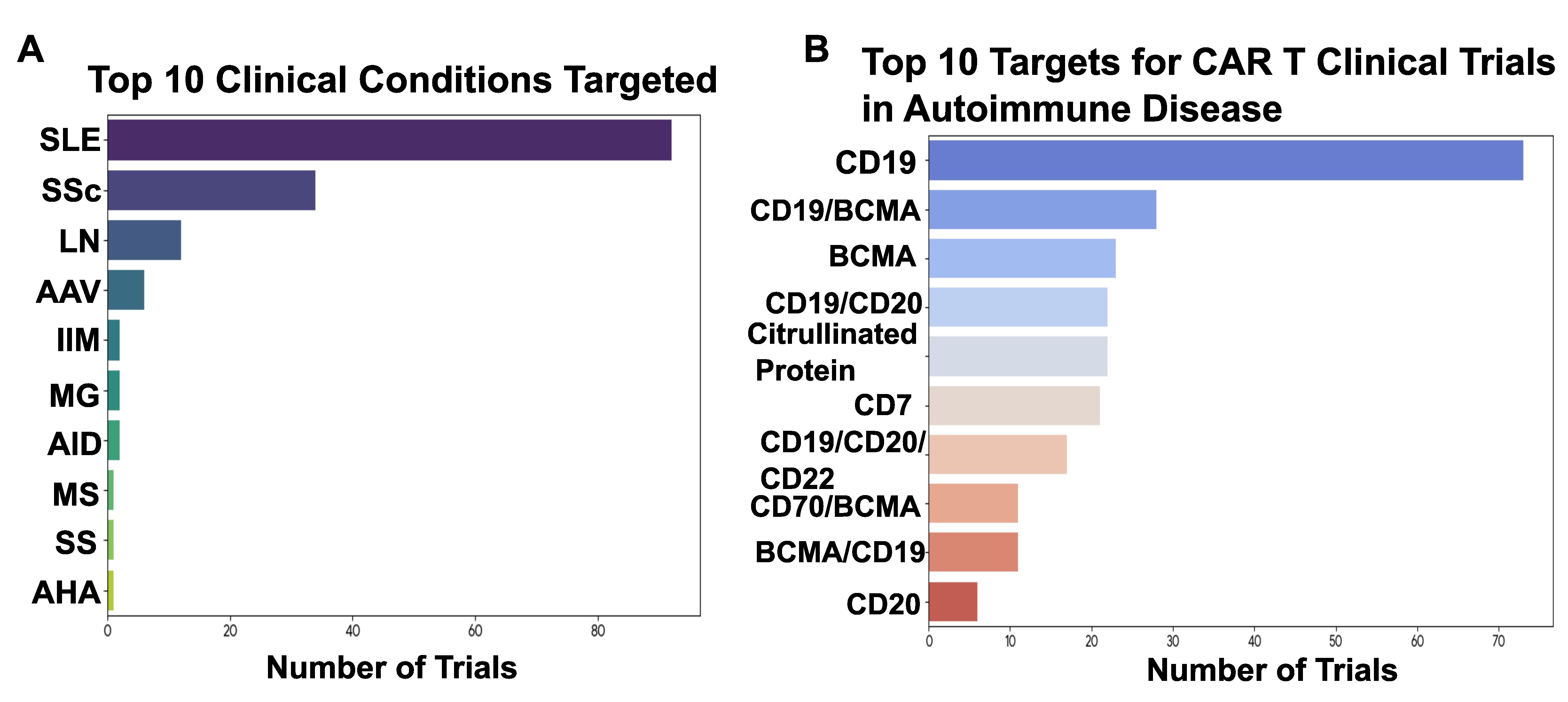
Figure 1. Clinical trials using traditional CAR T-cell therapy targeting B cells. A, A horizontal bar chart ranking autoimmune disorders by the number of active clinical trials. Systemic Lupus Erythematosus (SLE) represents the most frequently targeted condition, with significant activity also noted in Systemic Sclerosis (SSc), Lupus Nephritis (LN), and Myasthenia Gravis (MG). B, A bar chart illustrating the distribution of molecular targets utilized in current trials. CD19 is the most prevalent target, followed by dual-targeting strategies such as CD19 and BCMA. Other targets include BCMA alone and combinations involving CD20 and CD22. This figure was modified from Table 1 of reference #26.
Mechanistic Insights into CAAR-T Function
Immune synapse formation
Ellebrecht et al. demonstrated that CAAR-T activation recapitulates key features of canonical T-cell signaling architecture [6]. High-resolution imaging revealed receptor clustering, actin cytoskeletal reorganization, and exclusion of CD45 from the synaptic interface, findings consistent with the kinetic segregation model of T-cell activation. These observations indicate that CAAR signaling adheres to well-established biophysical principles governing receptor-mediated cytotoxicity.
Impact of soluble autoantibodies
A critical concern with antigen-based targeting is the potential for circulating autoantibodies to block engineered receptors. Given that PV patients harbor high titers of anti-Dsg3 IgG, receptor neutralization represents a plausible theoretical limitation. Unexpectedly, soluble autoantibodies produced heterogeneous effects. While partial inhibition of cytotoxic activity was observed under certain conditions, target cell killing remained largely preserved. In addition, soluble antibodies could promote CAAR-T activation and proliferation in specific experimental contexts. These findings suggest that affinity and binding kinetics play central roles in determining functional outcomes. Low-affinity interactions may be rapidly displaced during receptor engagement, whereas higher-affinity antibodies generate more stable receptor occupancy. Collectively, these dynamics indicate that circulating autoantibodies may not necessarily compromise therapeutic efficacy.
Determinants of cytotoxic specificity
CAAR-T cells exhibited robust cytotoxicity against BCR-expressing target cells while sparing keratinocytes expressing native Dsg3. Several structural and biophysical mechanisms are likely to contribute to this specificity. T-cell activation requires precise intermembrane spacing [24], whereas desmosomal architecture imposes geometric constraints that may limit synapse formation. Additionally, cadherin interactions typically have micromolar affinities, which are below the thresholds commonly required for CAR-mediated cytotoxicity. Finally, antigen truncation strategies, such as EC1–4 constructs, may reduce unintended ligand engagement while preserving pathogenic epitopes. These findings underscore the importance of structural immunology in engineering receptor specificity and safety.
Safety considerations
Off-target toxicity remains a major limitation of CAR-T therapy [25]. CAAR-T cells targeting a native epithelial antigen, therefore, present unique safety challenges. Experimental observations demonstrated an absence of keratinocyte cytotoxicity, with no detectable evidence of desmosome-related tissue injury [6]. Moreover, Fcγ receptor-mediated redirected killing was not observed. Together, these findings suggest that antigen-specific targeting can be achieved without disrupting physiological tissue architecture, highlighting a potentially favorable safety profile.
Therapeutic implications
The CAAR-T strategy potentially offers several advantages over conventional therapies [26,27]. Most importantly, this approach enables antigen-specific elimination of pathogenic B cells, thereby preserving protective humoral immunity. Such specificity may reduce the need for chronic immunosuppression while offering the possibility of durable remission.
Notably, limitations commonly encountered in oncology may be less problematic in autoimmune applications. Antigen escape mechanisms are biologically constrained when pathogenicity itself depends on antigen recognition. Furthermore, the relatively low frequency of autoreactive B cells [28] may mitigate the risk of excessive cytokine release. Downregulation of BCR expression, while theoretically possible, would likely compromise clone survival.
Broader relevance to autoimmune diseases
Many autoimmune disorders are driven by pathogenic autoantibodies directed against defined molecular targets. Given the central pathogenic role of B cells, therapies targeting antibody-producing B cells were explored as promising treatments for autoimmune diseases. Targeting B-cell subsets with traditional CAR T cells spans a broad spectrum of markers across B-cell development and maturation, including pro-B cells, pre-B cells, and plasma cells (Figure 2).
In this context of the CAAR T cells, it may also be generalizable to a broad spectrum of antibody-mediated diseases, including myasthenia gravis [29], membranous nephropathy [30], neuromyelitis optica, autoimmune encephalitis, bullous pemphigoid, and potentially type 1 diabetes. Antigen-specific targeting, therefore, introduces a scalable conceptual model for precision intervention in autoimmunity [26].
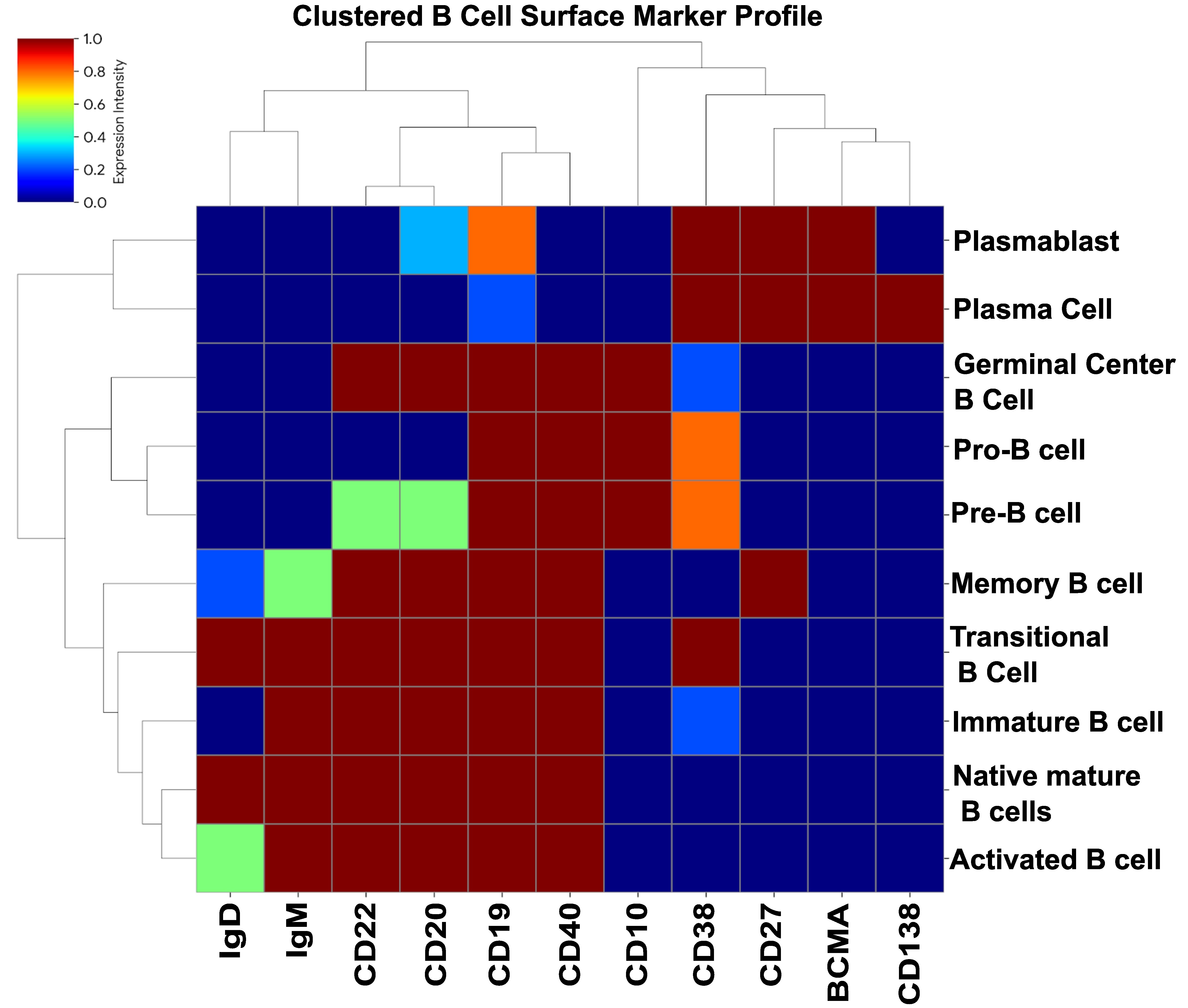
Figure 2. Cell surface markers expressed in B cell subsets. This heatmap illustrates the differential expression of key surface markers across various stages of B-cell development and maturation. The rows represent distinct B-cell subsets, including pro-B cells, pre-B cells, memory B cells, and plasma cells, while the columns display lineage-restricted markers such as CD19, CD20, CD22, and BCMA. The color gradient represents expression intensity, ranging from low/no expression (dark blue) to high expression (dark red).
Translational Challenges
Despite its promise, several challenges remain. First, CAAT therapies developed in the murine model using immune-deficient mice may show limited translatability to human autoimmune diseases. Moreover, additional pathogenic antibodies, including those directed against Dsg1 [31]and desmocollin 3 (Dsc3) [32], may also be present in the circulation of patients with pemphigus. Compared with T cells expressing CAAR (EC1–4), T cells expressing CAAR (EC1–5) demonstrated reduced efficacy. However, autoantibodies targeting EC5 have been reported to be pathogenic [33]. Moreover, using patient-derived autoreactive T cells, as detected in pemphigus [34], for the manufacturing of autologous CAAR T cells could potentially trigger disease flares. Key biological considerations include the persistence of CAAR-T, the contribution of long-lived plasma cells, epitope diversity, and potential compensatory immune mechanisms. Clinical implementation will require solutions to manufacturing complexity, cost, safety monitoring, and cytokine release management. Regulatory advancement will depend on standardizing antigen constructs, developing biomarkers, and optimizing patient selection criteria.
Future Directions
While numerous therapeutic strategies have been developed to target the B-cell compartment in pemphigus [5], specific and effective therapies that deplete pathogenic B cells remain to be developed. Dominant immunoglobulin heavy-chain variable region (VH) gene usage has been observed in several autoimmune diseases, such as VH1-46 in pemphigus and VH4-34 in SLE [35,36], highlighting a potential novel target for CAR T-cell therapy. Emerging opportunities include developing multi-antigen CAAR designs, receptor-affinity tuning strategies, incorporating safety switches, mRNA-based CAAR platforms, and combination immune-editing approaches. Integration with next-generation cellular engineering technologies may substantially enhance feasibility, safety, and therapeutic durability.
Conclusion
The CAAR-T cell strategy could represent a conceptual advance in autoimmune therapeutics by enabling antigen-specific immune editing, although clinical efficacy has yet to be demonstrated. Rather than suppressing immune responses or depleting entire cell lineages, this approach selectively eliminates pathogenic clones defined by antigen recognition. If successfully translated, CAAR-T cells may redefine treatment strategies for antibody-mediated autoimmune diseases, emphasizing precision, durability, and preservation of immune competence.
Fundings
This work is not funded.
Conflict of Interest
The authors have no conflicts to declare.
Authors Contribution
HY, manuscript drafting; LZ, data collection and figures; XM, manuscript editing.
References
2. Rosenblum MD, Remedios KA, Abbas AK. Mechanisms of human autoimmunity. J Clin Invest. 2015 Jun; 125(6): 2228–33.
3. Lallana EC, Fadul CE. Toxicities of immunosuppressive treatment of autoimmune neurologic diseases. Curr Neuropharmacol. 2011 Sep; 9(3): 468–77.
4. Zeng FAP, Wilson A, Sheriff T, Murrell DF. Side effects of steroid-sparing agents in patients with bullous pemphigoid and pemphigus: A systematic review. JAAD Int. 2022 Dec; 9:33–43.
5. Yuan H, Pan M, Chen H, Mao X. Immunotherapy for Pemphigus: Present and Future. Front Med (Lausanne). 2022 Jun 15;9:901239.
6. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. 2016 Jul 8;353(6295):179–84.
7. Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. 1991 Nov 29;67(5):869–77.
8. Amagai M, Stanley JR. Desmoglein as a target in skin disease and beyond. J Invest Dermatol. 2012 Mar;132(3 Pt 2):776–84.
9. Di Zenzo G, Amber KT, Sayar BS, Müller EJ, Borradori L. Immune response in pemphigus and beyond: progresses and emerging concepts. Semin Immunopathol. 2016 Jan;38(1):57–74.
10. Amagai M, Nishikawa T, Nousari HC, Anhalt GJ, Hashimoto T. Antibodies against desmoglein 3 (pemphigus vulgaris antigen) are present in sera from patients with paraneoplastic pemphigus and cause acantholysis in vivo in neonatal mice. J Clin Invest. 1998 Aug 15;102(4):775–82.
11. Aoki-Ota M, Tsunoda K, Ota T, Iwasaki T, Koyasu S, Amagai M, et al. A mouse model of pemphigus vulgaris by adoptive transfer of naive splenocytes from desmoglein 3 knockout mice. Br J Dermatol. 2004 Aug;151(2):346–54.
12. Payne AS, Ishii K, Kacir S, Lin C, Li H, Hanakawa Y, et al. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest. 2005 Apr;115(4):888–99.
13. Colliou N, Picard D, Caillot F, Calbo S, Le Corre S, Lim A, et al. Long-term remissions of severe pemphigus after rituximab therapy are associated with prolonged failure of desmoglein B cell response. Sci Transl Med. 2013 Mar 6;5(175):175ra30.
14. Hammers CM, Chen J, Lin C, Kacir S, Siegel DL, Payne AS, et al. Persistence of anti-desmoglein 3 IgG(+) B-cell clones in pemphigus patients over years. J Invest Dermatol. 2015 Mar;135(3):742–9.
15. Elmoursi A, Barmettler S. Therapeutic B-cell depletion: Mechanisms, clinical applications, and implications for secondary immunodeficiency. J Allergy Clin Immunol. 2025 Sep;156(3):597–603.
16. Lee J, Lundgren DK, Mao X, Manfredo-Vieira S, Nunez-Cruz S, Williams EF, et al. Antigen-specific B cell depletion for precision therapy of mucosal pemphigus vulgaris. J Clin Invest. 2020 Dec 1;130(12):6317–24.
17. Kasperkiewicz M, Ellebrecht CT, Takahashi H, Yamagami J, Zillikens D, Payne AS, et al. Pemphigus. Nat Rev Dis Primers. 2017 May 11;3:17026.
18. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013 Apr 18;368(16):1509–18.
19. Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014 Apr 10;123(15):2343–54.
20. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011 Aug 25;365(8):725–33
21. Mougiakakos D, Krönke G, Völkl S, Kretschmann S, Aigner M, Kharboutli S, et al. CD19-Targeted CAR T Cells in Refractory Systemic Lupus Erythematosus. N Engl J Med. 2021 Aug 5;385(6):567–9.
22. He X, Hu B, Zhang Y, Liu F, Li Q, Zheng C, et al. Treatment of two pediatric patients with refractory systemic lupus erythematosus using CD19-targeted CAR T-cells. Autoimmun Rev. 2025 Jan 3;24(1):103692.
23. Wang X, Wu X, Tan B, Zhu L, Zhang Y, Lin L, et al. Allogeneic CD19-targeted CAR-T therapy in patients with severe myositis and systemic sclerosis. Cell. 2024 Sep 5;187(18):4890–904.e9.
24. Cai H, Muller J, Depoil D, Mayya V, Sheetz MP, Dustin ML, et al. Full control of ligand positioning reveals spatial thresholds for T cell receptor triggering. Nat Nanotechnol. 2018 Jul;13(7):610–7.
25. Chohan KL, Siegler EL, Kenderian SS. CAR-T Cell Therapy: the Efficacy and Toxicity Balance. Curr Hematol Malig Rep. 2023 Apr;18(2):9–18.
26. Avouac J, Barzel A, Caiati D, Davis RS, Gottschalk S, Grieshaber-Bouyer R, et al. Roads and detours for CAR T cell therapy in autoimmune diseases. Nat Rev Drug Discov. 2026 Jan 26.
27. Vukovic J, Abazovic D, Vucetic D, Medenica S. CAR-engineered T cell therapy as an emerging strategy for treating autoimmune diseases. Front Med (Lausanne). 2024 Oct 10;11:1447147.
28. Pollmann R, Walter E, Schmidt T, Waschke J, Hertl M, Möbs C, et al. Identification of Autoreactive B Cell Subpopulations in Peripheral Blood of Autoimmune Patients With Pemphigus Vulgaris. Front Immunol. 2019 Jun 14;10:1375.
29. Oh S, Mao X, Manfredo-Vieira S, Lee J, Patel D, Choi EJ, et al. Precision targeting of autoantigen-specific B cells in muscle-specific tyrosine kinase myasthenia gravis with chimeric autoantibody receptor T cells. Nat Biotechnol. 2023 Sep;41(9):1229–38.
30. Altun B, Zhao G, Wang B, Mao X, Manfredo-Vieira S, Willis E, et al. Preclinical evaluation of antigen-specific B-cell depletion for phospholipase A2 receptor membranous nephropathy with chimeric autoantibody receptor T cells. Kidney Int. 2026 Jan;109(1):89–100.
31. Hammers CM, Stanley JR. Mechanisms of Disease: Pemphigus and Bullous Pemphigoid. Annu Rev Pathol. 2016 May 23;11:175–97.
32. Mao X, Nagler AR, Farber SA, Choi EJ, Jackson LH, Leiferman KM, et al. Autoimmunity to desmocollin 3 in pemphigus vulgaris. Am J Pathol. 2010 Dec;177(6):2724–30.
33. Hudemann C, Exner Y, Pollmann R, Schneider K, Zakrzewicz A, Feldhoff S, et al. IgG against the Membrane-Proximal Portion of the Desmoglein 3 Ectodomain Induces Loss of Keratinocyte Adhesion, a Hallmark in Pemphigus Vulgaris. J Invest Dermatol. 2023 Feb;143(2):254–63.e3.
34. Didona D, Scarsella L, Hudemann C, Volkmann K, Zimmer CL, Beckert B, et al. Type 2 T-Cell Responses against Distinct Epitopes of the Desmoglein 3 Ectodomain in Pemphigus Vulgaris. J Invest Dermatol. 2024 Feb;144(2):263–72.e8.
35. Cho MJ, Lo AS, Mao X, Nagler AR, Ellebrecht CT, Mukherjee EM, et al. Shared VH1-46 gene usage by pemphigus vulgaris autoantibodies indicates common humoral immune responses among patients. Nat Commun. 2014 Jun 19;5:4167.
36. Ramba M, Bogunovic D. Targeting autoreactive B cells in SLE with anti-9G4 synthetic immune receptor T cells. Nat Rev Immunol. 2026 Jan;26(1):5.