Commentary
When a patient is diagnosed with a glioblastoma, they are often blindsided with the diagnosis, experiencing symptoms typically developing over only days to weeks and then often undergoing a craniotomy within days of presentation, only to then be given a terminal diagnosis. Patients often ask “why me,” “what did I do to cause this to occur,” and “is this hereditary?”
Less than 5% of glioblastomas result from a hereditary syndrome. While not common, they do occur and perhaps may be under-recognized if family history is not known. Now, with more frequent germline testing done as a component of next generation tumor sequencing, it is hypothesized that these hereditary syndromes are better detected. This improved detection is not only beneficial for screening family members and screening the patient for other associated malignancies, but this opens up an opportunity for us as clinicians and scientists to better understand the tumorigenesis of glioblastoma in hereditary syndromes, which in turn may offer individualized treatment regimens.
The use of immunotherapy in glioblastoma has continued to present challenges despite its success in several malignancies. Immune checkpoint inhibitors have changed the treatment landscape for many cancers, including melanoma, renal cell carcinoma, head and neck squamous cell carcinoma, and others [1], but unfortunately have not shown the same results in glioblastoma. Checkmate 143 was a phase III clinical trial investigating nivolumab compared to bevacizumab at first recurrence in patients with glioblastoma. This study did not meet the primary end point of improved overall survival with median overall survival comparable in both arms [2]. This lack of success has been attributed to several factors specific to glioblastoma. While we now know the central nervous system is subject to active immunosurveillance and immune responses [3], glioblastoma has been proven to induce both local and systemic immunosuppression thereby evading the immune response. This appears to result from factors intrinsic to glioblastoma as well as factors related to the host response itself [4,5]. There are numerous ongoing efforts to overcome or circumvent the glioblastoma-induced immunosuppressive state through combinatory approaches of immunotherapy.
We recently reported the case report of a patient with Lynch Syndrome and glioblastoma treated with nivolumab, an immune checkpoint inhibitor [6]. Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer, is an autosomal dominant disorder caused by mutations in mismatch repair (MMR) genes. Patients with Lynch syndrome are at high risk of synchronous cancers, with the risk level dependent on which MMR mutation is harbored. Glioblastomas have a known association and occurrence with Lynch Syndrome, with an estimated risk of 1-6% of brain malignancy in patients with Lynch Syndrome [7]. However, the literature is sparse regarding incidence and null regarding unique treatments related to glioblastoma in the setting of Lynch Syndrome.
Immune checkpoint inhibition has proven great success initially in colon cancer with microsatellite instability (MSI) or mismatch repair deficiency (MMR-d), as in Lynch Syndrome [8], and later with other malignancies associated with the same features [9,10]. Its use however in glioblastoma as a whole, not selecting out for MSI or MMR-d, has been disappointing to say the least.
Our patient had a known diagnosis of Lynch Syndrome, diagnosed after a colon cancer diagnosis 10 years prior to her glioblastoma presentation. She was initially diagnosed with isocitrate dehydrogenase (IDH) wild type glioblastoma with O6-methylguanine- DNA methyltransferase (MGMT) unmethylation after craniotomy for tumor resection, subsequently receiving radiation with concurrent temozolomide, but was unable to receive adjuvant temozolomide due to cytopenia. She was treated with tumor treating fields for 18 months. She then had recurrence at a distal intracranial location for which she again underwent craniotomy for tumor resection, with pathology again consistent with IDH wild type, high grade glioma, MGMT unmethylated. Her tumor was found to be microsatellite unstable with a high tumor mutational burden. She was subsequently re-irradiated and treated with off label nivolumab which was continued after radiation completion without recurrent after now 40 months of nivolumab treatment. Additionally, she is without other systemic malignancy, including without recurrence of her more remote diagnosis of colon adenocarcinoma.
Anghileri et al. simultaneously published a similar case of a Lynch syndrome-associated recurrent glioblastoma responding to nivolumab. This patient did not have known Lynch syndrome at the time of glioblastoma diagnosis, but was discovered through germline sequencing revealing a pathogenic mutation in the MSH2 gene. Their patient was reported as still receiving nivolumab 68 months after recurrence [11]. They performed mutation signature analysis of both the initial and recurrent tumor, reporting that both specimens demonstrated mismatch repair deficiency. Additionally, they reported that the mutational burden in their case was “significantly higher than that in most hypermutant GBM patient data in TCGA”[11]. Both our case report and that of Anghileri et al. correlate response to nivolumab with tumor mutational burden.
This is being further investigated in the currently enrolling Alliance trial evaluating nivolumab and ipilimumab in patients with recurrent glioblastoma demonstrating elevated tumor mutational burden [NCT04145115].
Both case reports of IDH wild type, glioblastoma in the setting of Lynch Syndrome responding to nivolumab highlight the importance of an individualized approach in managing patients with glioblastoma. It appears that the microsatellite instability harbored in our patient’s glioblastoma correlates with the response to immune checkpoint inhibition with nivolumab. These cases shed light not only on the potential use of immune checkpoint inhibition in glioblastoma with tumor features, or genetic syndromes, associated with response to the same therapy for other malignancies, but more broadly shed light on the need to individualize glioblastoma treatment, particularly in the setting of glioblastoma as a result of an inherited syndrome.
Unique in our case report was the use of checkpoint inhibition concurrently with radiation therapy. The combination of immunotherapy with radiation therapy has been shown to potentially boost the abscopal effect seen in other malignancies treated with radiation therapy. The abscopal effect was first described in 1953 by Mole, where he described an immune-mediated response to radiation by distant tumor cells resulting from immune system activation by antigen release during radiotherapy (Figure 1) [12].
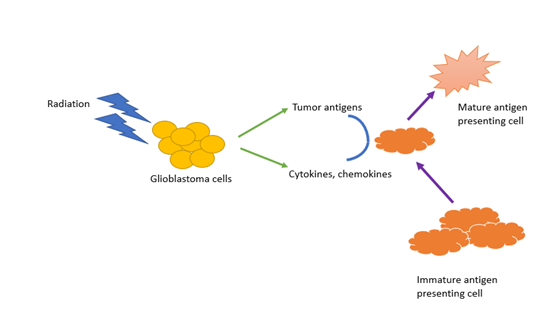
More recently, there is a growing body of evidence supporting the role of immunotherapy in boosting the abscopal effect [13]. This is particularly of interest in the field of glioblastoma given the infiltrative nature of the disease and the high rate of recurrence just outside the radiation field. Further research is needed to investigate the dynamics between immunotherapy and radiation therapy in glioblastoma treatment.
There have been improved collaborative efforts in glioblastoma, a designated rare disease. However, glioblastoma in the setting of Lynch Syndrome represents an even rarer diagnosis necessitating international collaboration and perhaps a consortium effort to further investigate and treat glioblastoma in this setting. In writing our case report, we are putting forward an ask to researchers and clinicians to join in the effort to individualize therapy. With the changes first in 2016 to the World Health Organization Central Nervous System Tumor Classification and the more recent 2021 WHO update, the door to understand the variability of glioblastoma has been opened which will hopefully lead to tailored therapies such as we report in our Lynch Syndrome patient without progression on immunotherapy for her recurrent IDH wild type glioblastoma.
References
2. Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the CheckMate 143 phase 3 randomized clinical trial. JAMA Oncology. 2020 Jul 1; 6(7):1003-10.
3. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015 Jul; 523(7560):337-41.
4. Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nature reviews Clinical Oncology. 2018 Jul; 15(7):422-42.
5. Roszman T, Elliott L, Brooks W. Modulation of T-cell function by gliomas. Immunology today. 1991 Oct 1; 12(10):370-4.
6. Sherman WJ, Vitaz TW. Nivolumab with radiation therapy in a glioblastoma patient with Lynch syndrome. BMJ Case Reports CP. 2021 Apr 1; 14(4):e241026.
7. Barrow E, Hill J, Evans DG. Cancer risk in Lynch syndrome. Familial Cancer. 2013 Jun 1; 12(2):229-40.
8. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. The Lancet Oncology. 2017 Sep 1; 18(9):1182-91.
9. Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord JP, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair–deficient cancer: Results from the phase II KEYNOTE-158 study. Journal of Clinical Oncology. 2020 Jan 1; 38(1):1.
10. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatchrepair deficiency. New England Journal of Medicine. 2015 Jun 25; 372(26):2509-20.
11. Anghileri E, Di Ianni N, Paterra R, Langella T, Zhao J, Eoli M, et al. High tumor mutational burden and T-cell activation are associated with long-term response to anti- PD1 therapy in Lynch syndrome recurrent glioblastoma patient. Cancer Immunology, Immunotherapy. 2021 Mar; 70(3):831-42.
12. Mole RH. Whole body irradiation—radiobiology or medicine?. The British Journal of Radiology. 1953 May; 26(305):234-41.
13. Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC. Using immunotherapy to boost the abscopal effect. Nature Reviews Cancer. 2018 May; 18(5):313-22.