Abstract
Mutations in a single TRPV6 allele trigger chronic pancreatitis while loss of both alleles is responsible for skeletal dysplasia in newborns. The latter clinical presentation is accompanied by elevated serum levels of the parathyroid hormone, a condition known as transient neonatal hyperparathyroidism (TNHP). In humans, TRPV6 is abundantly expressed in the primary fetal-maternal interface in the placenta, as well as in pancreatic acini and within a few exocrine glands including salivary and lacrimal glands. However, inconsistent results regarding the cellular localization have complicated elucidation of the exact function within these tissues. We have verified an intracellular localization of TRPV6 within vesicles and identified a sorting motive, a glycosylation site and an ER-retention motive within the sequence, which together account for the observed channel localization. This is in contradiction to postulated functions of TRPV6 being responsible for calcium uptake into epithelial cells, which would require TRPV6 localization within the plasma membrane of expressing cells. We suggest that previous observations which demonstrate TRPV6 expression at the plasma membrane may result from overexpression and do not represent endogenous channel positioning. We showed that normal TRPV6 function requires the action of an enzyme (GNPTAB) and the subsequent interaction with a mannose-6-phosphate receptor to be delivered to endosomes. The GNPTAB enzyme marks proteins with mannose-6-phosphate and is known to be defective in patients suffering from mucolipidosis type II, which also present with skeletal dysplasia with elevated parathyroid hormone. The reliance of TRPV6 on this enzyme for correct localization and therefore function provides a possible explanation why patients suffering from either mucolipidosis type II or TRPV6 malfunction exhibit overlapping symptoms.
Keywords
Ion channel, Mucolipidosis type II, Chronic pancreatitis, Skeletal dysplasia
Special Features of TRPV6
- Highly calcium-selective channel
- Different expression pattern between humans and rodents
- Coupled polymorphisms in humans differentiate two TRPV6 alleles (TRPV6a, TRPV6b)
- Strong selective pressure on the TRPV6b variant (found exclusively in humans)
- N-terminal extension of the TRPV6 protein only in placental animals (TRPV6 expressed in placenta)
- Translation initiated at an ACG triplet
- The initiation triplet (ACG) translated as methionine
- ACG serves as TRPV6 initiation triplet in mammals, except in flying foxes and bats
- N-terminal coding region of the mRNA forms stable stem loop which decreases translation efficiency of the TRPV6 protein
- Contains an atypical ER retention motive
- Overexpressed in prostate, breast and endometrial cancer
- Mutation in a single allele causes chronic pancreatitis (TRPV6 expressed in pancreatic acinus cells)
- Mutation in both alleles causes skeletal dysplasia with increased level of the parathyroid hormone
- Link between TRPV6 and mucolipidosis type II
Commentary
Since the cloning of the TRPV6 cDNA about 25 years ago from rodents and humans [1–4], it has become clear that TRPV6 possesses a few unique features. The family of TRP-channels consists of 28 members [5], most of which are non-selective cation-conducting channels. In contrast, TRPV6 (and TRPV5) is highly selective for calcium [3,6], a property which has been conserved throughout evolution. Evolutionarily old organisms such as echinoderms (starfishes and sea urchins), which are known to have developed more than 600 million years ago [7] have also retained this calcium-selective pore. Using extrapolation and molecular clock mapping, the age of the TRPV6 ancestral gene has been estimated at 730 million years with a relatively constant mutation rate of one amino acid per million years. This has led to a lower sequence identity between human and rodent TRPV6 (~90%) than is observed for other TRP genes such as TRPC4 [7].
Mammalian TRPV6 and TRPV5 most likely arose by gene duplication from a common ancestor and as consequence, the genes are localized in tandem on the human chromosome 7q33-34 [2]. TRPV5 is predominantly expressed in epithelial cells of the distal convolute of the kidney where it functions as a calcium re-uptake channel. It has been speculated that the appearance of TRPV5 is connected to the evolution of organisms with a second kidney [8,9], however, TRPV5-like genes can be found in lancelets which developed more than 500 million years ago [7]. Over time, partially loss or doubling of genetic material during evolution has led to organisms with varied numbers of TRPV5 and TRPV6 genes. Birds, for example, have completely lost the TRPV6 gene whereas salmonid fish species possess two TRPV6 copies. A complex model of early doubling events of TRPV5 and TRPV6 genes has been postulated by Flores-Aldama and co-workers [10].
A surprising observation has been described exclusively in placental animals; the TRPV6 protein contains an N-terminal extension of 40 amino acids, corresponding to 120 bases within the mRNA. This relatively rare mechanism does not influence the function of the channel, but greatly reduces the translation effectiveness of the TRPV6 mRNA [11]. Translation is initiated from an ACG triplet at the beginning of this extension which is translated as methionine. Despite the continued presence of an in-frame AUG located downstream in most mammals, we have speculated that the initiator tRNA-Met is placed on the noncanonical ACG codon due to presence of a stabile stem loop structure which prevents the translational machinery from using the standard AUG codon [12]. As overexpression of TRPV6 in conventional cell lines is known to cause dramatic calcium overload, this reduced translation is likely an evolutionary mechanism evolved to prevent toxic calcium overload within the cytosol of TRPV6-expressing cells [13]. Intriguingly, while almost all placental animals initiate translation from this ACG triplet, this is not the case in flying foxes and bats. These animals contain instead a classical AUG triplet at the position corresponding to the upstream ACG codon. Overexpression experiments using chimeras of human and bat TRPV6 show that this modification is responsible for significantly enhanced protein synthesis in these species, speculated to be required to compensate for limited dietary calcium within these animals, especially during pregnancy [12]. One unique mammal, the duck-billed platypus, which does not have a placenta and still lays eggs, possesses only a part of the N-terminal extension, arguing for a role of the extended TRPV6 sequence in placental function [7].
In humans, TRPV6 is abundantly expressed in the placental syncytiotrophoblast – the primary fetal-maternal interface – as well as in pancreatic acini and within a few exocrine glands including salivary and lacrimal glands. Evolutionarily obtained polymorphisms of the TRPV6 gene have been described exclusively in humans, giving rise to two alleles of the TRPV6 gene. Five base changes affecting three amino acid positions within TRPV6 cDNA (C157R, M378V, M681T) have been identified which seem to be genetically linked, leading to an older TRPV6 isoform termed TRPV6a (R157, V378, T681) and a modern variant termed TRPV6b (C157, M378, M681) [3]. The TRPV6a variant is still abundantly present in South-African population (up to 80% of individuals homozygous TRPV6a) whereas the TRPV6b allele is dominant with increasing distance to the African continent (up to 96% homozygous TRPV6b) [14]. Although TRPV6a and TRPV6b channels exhibit similar channel properties [15], the authors suggested that selection pressure may lead to this isoform distribution, but what might drive this selection is still unknown.
Aberrant TRPV6 expression has been connected to several diseases. Carcinomas of the prostate, breast and endometrium overexpress TRPV6 transcripts, whereas the corresponding healthy tissues do not [3,16–22]. In the case of prostate cancer, it was demonstrated that TRPV6 expression is correlated with the malignancy of the cancer cells. Thus, TRPV6-positive tumors were more likely to metastasize beyond the prostate capsule. Based on these data, it was concluded that TRPV6 is a driver for metastatic properties of several malignancies.
Another disease associated with abnormal TRPV6 expression was found to be non-alcohol-dependent chronic pancreatitis [23–27]. Whole exon sequencing revealed several mutations of the TRPV6 gene, which, when overexpressed, dramatically reduced channel activity (<50%). The underlying mechanism for development of this disease is not known but likely reflects disruption of normal TRPV6 function in pancreatic acinus cells. Of note, only one TRPV6 allele was typically mutated in these patients whereas the second allele was still intact.
Patients with mutations on both alleles present a reduced calcification of the skeleton at birth accompanied with increased serum levels of the parathyroid hormone [28–32]. The skeletal dysplasia observed in these patients most likely results from reduced calcium transport through the placenta during the pregnancy coupled with compensatory upregulation of the parathyroid hormone. The mutations identified in these patients typically affect the conductivity of the TRPV6 channel [29]. Since TRPV6 is expressed in the syncytiotrophoblast layer of the placenta, the logical conclusion is that calcium is transported through TRPV6 channels directly. This idea was supported by the finding that TRPV6 overexpression in all cell lines used to date results in calcium influx across the plasma membrane, which could be easily demonstrated by patch clamp or calcium imaging. However, in tissues or cell lines which endogenously express TRPV6, endogenous TRPV6 currents could not be found. TRPV6-like currents have never been measured in syncytiotrophoblast cells and are in addition not detectable in a trophoblast-derived cell line, BeWo, which expresses TRPV6. Also, in two cancer-derived cell lines, T47D and LNCaP, which express the TRPV6-mRNA, no TRPV6 currents could be measured. In addition, HEK293 cells which overexpress a fluorescently-tagged-TRPV6 fusion construct, have no detectable staining at the plasma membrane [11].
Based upon these inconsistent results, we focused on the intracellular destination of TRPV6 and could show that the channel is indeed transported to intracellular vesicles [33]. Firstly, microscopic analysis following transfection of TRPV6 fused to either GFP or RFP confirmed vesicular staining and an absence of signal within the plasma membrane or filopodia. Merged microscopic pictures of TRPV6-GFP and TRPV6-RFP showed that identical vesicles were labelled and staining with Rab5 confirmed these vesicles as early endosomes [34]. Surprisingly, TRPV6 currents were measurable in these cells by patch clamp and calcium imaging, indicating that a small proportion of the TRPV6 protein must reach the plasma membrane under these conditions. Comparative measurements of TRPV6 fused to GFP and wild type TRPV6 cloned in an IRES-GFP plasmid (not fused to the GFP cDNA) showed identical TRPV6 activity and kinetics, indicating that the fusion of GFP or RFP does not influence the activity of TRPV6. In addition, we could show that neither fluorescence tag influenced the intracellular trafficking of TRPV6. We stained pancreatic acini and placenta syncytiotrophoblast with TRPV6-specific antibodies and confirmed the presence of TRPV6-positive vesicles, whereas plasma membrane signal was again not detectable.
A N-glycosylation site (N398), located between the first and second transmembrane domains of TRPV6, is responsible for vesicular transport of TRPV6. To determine this, the localization of the closely related TRPV4 channel, which does not possess an N-glycosylation site at the corresponding position was studied. Overexpression of the TRPV4-GFP fusion construct in cells led to reliable detection of signal within the plasma membrane as well as measurement of TRPV4 currents. A TRPV4 chimera which included the N-glycosylation site of TRPV6 was redirected to intracellular vesicles. When the critical amino acid responsible for the N-glycosylation, asparagine (N), was substituted to alanine in the chimera, this second chimera was localized back in the plasma membrane. These experiments showed that the N-glycosylation site of TRPV6 is responsible for directing the TRPV4-V6 chimera into vesicles and further, that this site underlies vesicular transport of TRPV6.
Interestingly, when the asparagines of the N-glycosylation site (N397A, N398A) in TRPV6 were substituted to alanine, this construct was found exclusively in ER membranes of overexpressing cells. This is in contrast to the localization of TRPV4 which, without a N-glycosylation site, is delivered to the plasma membrane. Therefore, we questioned, whether the TRPV6 sequence contains an ER-retention motive responsible for retaining TRPV6 mutant in the ER. Indeed, TRPV6 contains a C-terminal located RDEL motive, similar to the classical ER retention motive present in soluble ER-proteins (KDEL), within the TRPV6 sequence. A mutation of this motive (RAEL) resulted in plasma membrane staining within TRPV6-RAEL-expressing cells, indicating that this sequence functions as an ER retention signal. This RDEL sequence is located in close proximity to the highly conserved TRP-Box of TRP channels, but is only present in TRPV6 and TRPV5 and none of the other TRP channels. As a consequence, the calcium uptake of TRPV6-RAEL-expressing cells was increased compared to wild type TRPV6.
As described above, patients with one defective TRPV6 allele develop chronic pancreatitis whereas patients with two defective alleles develop a skeletal dysplasia with elevated serum parathyroid levels. In general, TRPV6 mutations have been shown to decrease channel activity to <50%. However, one mutation frequently found in Japanese pancreatitis patients, I223T, still retains normal channel activity [23,33]. Suzuki and co-workers reported a family in which a child carrying the I223T mutation alongside a second TRPV6 mutation R425Q [29] was born with skeletal dysplasia. This raised the question why this seemingly fully-functional channel still manifested in pancreatitis and skeletal dysplasia in these patients. Using TRPV6 containing the R425Q mutation reduced channel activity but co-expression of both cDNAs (TRPV6-I223T and TRPV6-R425Q) demonstrated that the I223T-construct could compensate for the second mutation, fully restoring TRPV6 activity in this scenario [33]. However, we found the TRPV6-I223T-channel is retained in the ER and not transferred to vesicles due to disruption of a sorting motive. We assume that the I223T mutation drives both diseases, chronic pancreatitis and skeletal dysplasia (in combination with the second mutation), due to incorrect localization of TRPV6. We also tested another mutation, G428R, which significantly reduces the TRPV6-activity [29] and co-expressed this mutant with TRPV6-I223T. The decreased activity of the G428R mutation could also be compensated by the I223T mutation (unpublished).
Several case reports show that patients suffering from mucolipidosis type II exhibit overlapping symptoms with TRPV6 deficiency, namely skeletal dysplasia accompanied with elevated parathyroid hormone in the serum at the time of birth [35,36]. Mucolipidosis type II patients exhibit defects in an enzyme (GNPTAB, N-Acetylglucosamine-1-phosphate transferase, [37,38]). This enzyme marks proteins at N-glycosyl side chains with mannose-6-phosphate. Proteins which are mannosylated in this way interact with a mannose-6-phosphate receptor and are delivered to endosomes/lysosomes. It could be shown that TRPV6 interacts with the mannose-6-phosphat receptor (CI-MPR, IGF2R), indicating that TRPV6 is likely mannosylated at the glycosyl side chain and targeted to endosomes. This is consistent with localization experiments showing vesicular staining using a fusion construct of TRPV6-GFP which colocalizes with the endosomal marker Rab5 (Figure 1).
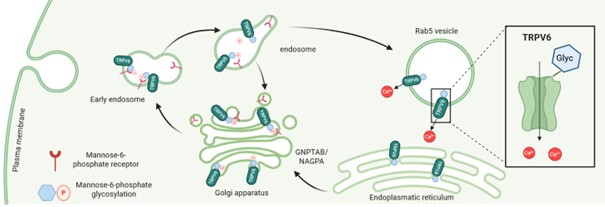
Figure 1. The glycosylation site (N398) is responsible for vesicular localization. The TRPV6 protein is glycosylated and mannosylated with mannose-6-phosphate within the Golgi apparatus and after interaction with a mannose-6-phosphate receptor (CI-MPR/IGF2R) transported to endosomes. TRPV6 is predominantly found in Rab5-positive vesicles (created with Biorender.com).
The α/β -subunits of GNPTAB are translated from one gene and cleaved by a S1-protease into two independent subunits, a requisite step in generating an active GNPTAB enzyme. Following incubation with an inhibitor of the S1-protease, PF-429242 [39], TRPV6 expression at the plasma membrane increased, supporting a role for GNPTAB in vesicular localization of TRPV6 within the endosome. If the N-glycosylation is completely blocked (by mutation) TRPV6 is retained in the ER. But blocking only the mannosylation leads to increased TRPV6 in the plasma membrane (Figures 2 and 3). These results indicate that there is a crosstalk between TRPV6 and GNPTAB and might explain why patients show overlapping symptoms observed in both mucolipidosis type II and TNHP (transient neonatal hyperparathoidism) driven by TRPV6 deficiency.
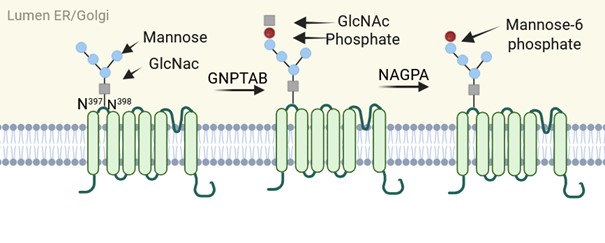
Figure 2. TRPV6 glycosylation occurs at asparagine N398. Through the action of GNPTAB and NAGPA, the glycosyl side chain is labelled with mannose-6-phosphate. GNPTAB consists of α2β2γ2-subunits. For generation of an active GNPTAB enzyme, the cleavage of ab precursor by site1-protease (S1-protease) is required. The activity of GNPTAB can be minimized by blocking the S1-protease with inhibitor PF-429242 (created with Biorender.com).
Based on the results described, TRPV6 is probably an intracellular channel which is endogenously localized within endosomes. We have identified three sequence motives that are thought to prevent TRPV6 from being transported to the plasma membrane (Figure 3). We show that mutations within these motives influence the trafficking of the TRPV6 channel but not the function in terms of calcium permeability. Thus, TRPV6-dependent plasma membrane currents may result from spillover effect caused by overexpression, providing an explanation for the inconsistent results described to date. This then raises questions about the actual function of TRPV6 channels within cells. Stainings of placenta syncytiotrophoblast and pancreatic acini with TRPV6 specific antibodies indicate vesicular localization of the channel, which would by definition preclude a role for this channel in the direct transport of calcium, as was supposed [8,40,41]. In addition, low TRPV6 protein levels make it is difficult to imagine how the large amounts of calcium required during late pregnancy (up to 300 mg/day [42]) could be transported via TRPV6 without measurable currents being detectable in the syncytiotrophoblast. However, patients born with skeletal dysplasia as a result of mutated TRPV6 show normalization later in life upon calcium supplementation in the diet, supporting that TRPV6 disruption does indeed perturb transplacental calcium transport in some way but uptake is unaffected [28,43].
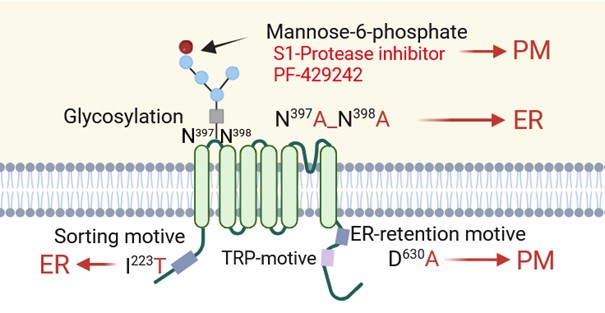
Figure 3. Mutations which affect the localization of TRPV6. A mutation of the sorting motive, I223T, and of the glycosylation site, N397A, N398A, prevent TRPV6 from being transported out of the endoplasmic reticulum. The amount of TRPV6 in the plasma membrane (PM) increases if only the mannosylation with mannose-6-phosphate is blocked by S1-protease inhibitor, PF-429242, or by mutation of the ER-retention motive (D630A) (created with Biorender.com).
In any case, an active uptake system for calcium ions exists in both the small intestine and the syncytiotrophoblast; furthermore, the first TRPV6 mRNA was isolated from the rat small intestine using a functional cloning approach. The expression data, taken together with the properties of the overexpressed channel, were reasonably interpreted to suggest that TRPV6 is a suitable candidate for this function [1,16,44, 45], namely calcium uptake in epithelial cells. The fact that dysfunctional TRPV6 channels lead to skeletal abnormalities in both KO mice and humans also supports the notion that TRPV6 is involved in calcium transport across the placenta [46]. Additionally, TRPV6 expression is found in murine uterine epithelial cells (MEECs) [47]. In these cells, however, only under divalent-free conditions can a very small, TRPV6-mediated transmembrane sodium influx be detected (~2 pA/pF at -80 mV). Since the conductance for Ca2+ is approximately 10 times lower than for Na+ [3], it can be concluded that even in MEECs, TRPV6 contributes only insignificantly to calcium uptake into these cells. In contrast, intracellular calcium oscillations are significantly reduced in KO cells. Taken together, the results imply that TRPV6 is primarily involved in intracellular calcium signaling and may play only a minor role in the plasma membrane.
TRPV6 expression is observed in glandular tissues (salivary-, lacrimal-gland and pancreatic acinus cells) leading to the speculation that channel function could be connected to secretion processes in these tissues. This finding is supported by TRPV6 mutations which trigger chronic pancreatitis, a disease which occurs if the secretion of zymogens is affected. The syncytiotrophoblast also exhibits features similar to glandular tissues and secretes several hormones which drive placenta differentiation. Thus, if placental differentiation is affected this could alter the surface of the syncytiotrophoblast, thereby influencing transcellular and paracellular Ca2+ transport.
In summary we suggest that TRPV6 is involved in the secretory function of these organs and is not primarily involved in Ca2+ uptake in epithelial cells. In this respect, the observable deficits in transplacental calcium transport are likely to be attributable to indirect effects.
References
2. Peng JB, Chen XZ, Berger UV, Weremowicz S, Morton CC, Vassilev PM, et al. Human calcium transport protein CaT1. Biochem Biophys Res Commun. 2000 Nov 19;278(2):326–32.
3. Wissenbach U, Niemeyer BA, Fixemer T, Schneidewind A, Trost C, Cavalie A, et al. Expression of CaT-like, a novel calcium-selective channel, correlates with the malignancy of prostate cancer. J Biol Chem. 2001 Jun 1;276(22):19461–8.
4. Hirnet D, Olausson J, Fecher-Trost C, Bödding M, Nastainczyk W, Wissenbach U, et al. The TRPV6 gene, cDNA and protein. Cell Calcium. 2003 May-Jun;33(5-6):509–18.
5. Montell C, Birnbaumer L, Flockerzi V. The TRP channels, a remarkably functional family. Cell. 2002 Mar 8;108(5):595–8.
6. Voets T, Janssens A, Prenen J, Droogmans G, Nilius B. Mg2+-dependent gating and strong inward rectification of the cation channel TRPV6. J Gen Physiol. 2003 Mar;121(3):245–60.
7. Wolske K, Wyatt A, Wissenbach U. Comments on the evolution of TRPV6. Ann Anat. 2021 Nov;238:151753.
8. Peng JB. TRPV5 and TRPV6 in transcellular Ca(2+) transport: regulation, gene duplication, and polymorphisms in African populations. Adv Exp Med Biol. 2011;704:239–75.
9. Peng JB, Chen XZ, Berger UV, Vassilev PM, Brown EM, Hediger MA. A rat kidney-specific calcium transporter in the distal nephron. J Biol Chem. 2000 Sep 8;275(36):28186–94.
10. Flores-Aldama L, Vandewege MW, Zavala K, Colenso CK, Gonzalez W, Brauchi SE, et al. Evolutionary analyses reveal independent origins of gene repertoires and structural motifs associated to fast inactivation in calcium-selective TRPV channels. Sci Rep. 2020 May 26;10(1):8684.
11. Fecher-Trost C, Wissenbach U, Beck A, Schalkowsky P, Stoerger C, Doerr J, et al. The in vivo TRPV6 protein starts at a non-AUG triplet, decoded as methionine, upstream of canonical initiation at AUG. J Biol Chem. 2013 Jun 7;288(23):16629–44.
12. Wolske K, Fecher-Trost C, Wesely C, Löhr H, Philipp S, Belkacemi A, et al. Why endogenous TRPV6 currents are not detectable-what can we learn from bats? Cell Calcium. 2020 Dec;92:102302.
13. Singh AK, McGoldrick LL, Twomey EC, Sobolevsky AI. Mechanism of calmodulin inactivation of the calcium-selective TRP channel TRPV6. Sci Adv. 2018 Aug 15;4(8):eaau6088.
14. Akey JM, Swanson WJ, Madeoy J, Eberle M, Shriver MD. TRPV6 exhibits unusual patterns of polymorphism and divergence in worldwide populations. Hum Mol Genet. 2006 Jul 1;15(13):2106–13.
15. Hughes DA, Tang K, Strotmann R, Schöneberg T, Prenen J, Nilius B, et al. Parallel selection on TRPV6 in human populations. PLoS One. 2008 Feb 27;3(2):e1686.
16. Zhuang L, Peng JB, Tou L, Takanaga H, Adam RM, Hediger MA, et al. Calcium-selective ion channel, CaT1, is apically localized in gastrointestinal tract epithelia and is aberrantly expressed in human malignancies. Lab Invest. 2002 Dec;82(12):1755–64.
17. Peng JB, Zhuang L, Berger UV, Adam RM, Williams BJ, Brown EM, et al. CaT1 expression correlates with tumor grade in prostate cancer. Biochem Biophys Res Commun. 2001 Apr 6;282(3):729–34.
18. Fixemer T, Wissenbach U, Flockerzi V, Bonkhoff H. Expression of the Ca2+-selective cation channel TRPV6 in human prostate cancer: a novel prognostic marker for tumor progression. Oncogene. 2003 Oct 30;22(49):7858–61.
19. Wissenbach U, Niemeyer B, Himmerkus N, Fixemer T, Bonkhoff H, Flockerzi V. TRPV6 and prostate cancer: cancer growth beyond the prostate correlates with increased TRPV6 Ca2+ channel expression. Biochem Biophys Res Commun. 2004 Oct 1;322(4):1359–63.
20. Wissenbach U, Niemeyer BA, Flockerzi V. TRP channels as potential drug targets. Biol Cell. 2004 Feb;96(1):47–54.
21. Bolanz KA, Hediger MA, Landowski CP. The role of TRPV6 in breast carcinogenesis. Mol Cancer Ther. 2008 Feb;7(2):271–9.
22. Bolanz KA, Kovacs GG, Landowski CP, Hediger MA. Tamoxifen inhibits TRPV6 activity via estrogen receptor-independent pathways in TRPV6-expressing MCF-7 breast cancer cells. Mol Cancer Res. 2009 Dec;7(12):2000–10.
23. Masamune A, Kotani H, Sörgel FL, Chen JM, Hamada S, Sakaguchi R, et al. Variants That Affect Function of Calcium Channel TRPV6 Are Associated With Early-Onset Chronic Pancreatitis. Gastroenterology. 2020 May;158(6):1626–41.e8.
24. Hamada S, Masson E, Chen JM, Sakaguchi R, Rebours V, Buscail L, et al. Functionally deficient TRPV6 variants contribute to hereditary and familial chronic pancreatitis. Hum Mutat. 2022 Feb;43(2):228–239.
25. Shah IA, Prasad H, Banerjee S, Kurien RT, Chowdhury SD, Visweswariah SS. A novel frameshift mutation in TRPV6 is associated with hereditary pancreatitis. Front Genet. 2023 Jan 9;13:1058057.
26. Goma M, Hagiwara SI, Wada T, Maeyama T, Okamoto N, Ishii S, et al. A case of early-onset idiopathic chronic pancreatitis associated with a loss-of-function TRPV6 p.R483Q variant successfully treated by pancreatic duct stenting. Clin J Gastroenterol. 2023 Aug;16(4):623–27.
27. Masamune A, Masson E, Zou WB, Rygiel AM, Chowdhury SD, Kikuta K, et al. TRPV6-related pancreatitis: natural history and the impact of the pancreas-specific deletion on pancreatitis in mice. J Gastroenterol. 2026 Feb;61(2):207–21.
28. Burren CP, Caswell R, Castle B, Welch CR, Hilliard TN, Smithson SF, et al. TRPV6 compound heterozygous variants result in impaired placental calcium transport and severe undermineralization and dysplasia of the fetal skeleton. Am J Med Genet A. 2018 Sep;176(9):1950–55.
29. Suzuki Y, Chitayat D, Sawada H, Deardorff MA, McLaughlin HM, Begtrup A, et al. TRPV6 Variants Interfere with Maternal-Fetal Calcium Transport through the Placenta and Cause Transient Neonatal Hyperparathyroidism. Am J Hum Genet. 2018 Jun 7;102(6):1104–14.
30. Yamashita S, Mizumoto H, Sawada H, Suzuki Y, Hata D. TRPV6 Gene Mutation in a Dizygous Twin With Transient Neonatal Hyperparathyroidism. J Endocr Soc. 2019 Jan 3;3(3):602–06.
31. Mason AE, Grier D, Smithson SF, Burren CP, Gradhand E. Post-mortem histology in transient receptor potential cation channel subfamily V member 6 (TRPV6) under-mineralising skeletal dysplasia suggests postnatal skeletal recovery: a case report. BMC Med Genet. 2020 Mar 30;21(1):64.
32. Almidani E, Elsidawi W, Almohamedi A, Bin Ahmed I, Alfadhel A. Case Report of Transient Neonatal Hyperparathyroidism: Medically Free Mother. Cureus. 2020 Feb 15;12(2):e7000.
33. Fecher-Trost C, Gehl AL, Trunk A, Hellmich J, Wesely C, Löhr H, et al. The intracellular localization and the ionic permeation of TRPV6 triggers chronic pancreatitis, skeletal dysplasia and is connected to mucolipidosis type II. Cell Commun Signal. 2025 Dec 23;24(1):44.
34. Kogel A, Fecher-Trost C, Wissenbach U, Flockerzi V, Schaefer M. Ca2+ transport via TRPV6 is regulated by rapid internalization of the channel. Cell Calcium. 2022 Sep;106:102634.
35. Heo JS, Choi KY, Sohn SH, Kim C, Kim YJ, Shin SH, et al. A case of mucolipidosis II presenting with prenatal skeletal dysplasia and severe secondary hyperparathyroidism at birth. Korean J Pediatr. 2012 Nov;55(11):438–44.
36. Boruah R, Monavari AA, Conlon T, Murphy N, Stroiescu A, Ryan S, et al. Secondary Hyperparathyroidism in Children with Mucolipidosis Type II (I-Cell Disease): Irish Experience. J Clin Med. 2022 Mar 2;11(5):1366.
37. Tiede S, Storch S, Lübke T, Henrissat B, Bargal R, Raas-Rothschild A, et al. Mucolipidosis II is caused by mutations in GNPTA encoding the alpha/beta GlcNAc-1-phosphotransferase. Nat Med. 2005 Oct;11(10):1109–12.
38. Tiede S, Cantz M, Spranger J, Braulke T. Missense mutation in the N-acetylglucosamine-1-phosphotransferase gene (GNPTA) in a patient with mucolipidosis II induces changes in the size and cellular distribution of GNPTG. Hum Mutat. 2006 Aug;27(8):830–1.
39. Hay BA, Abrams B, Zumbrunn AY, Valentine JJ, Warren LC, Petras SF, et al. Aminopyrrolidineamide inhibitors of site-1 protease. Bioorg Med Chem Lett. 2007 Aug 15;17(16):4411-4.
40. Stewart JM. TRPV6 as A Target for Cancer Therapy. J Cancer. 2020 Jan 1;11(2):374-87.
41. den Dekker E, Hoenderop JG, Nilius B, Bindels RJ. The epithelial calcium channels, TRPV5 & TRPV6: from identification towards regulation. Cell Calcium. 2003 May-Jun;33(5-6):497-507.
42. Kovacs CS. Maternal Mineral and Bone Metabolism During Pregnancy, Lactation, and Post-Weaning Recovery. Physiol Rev. 2016 Apr;96(2):449-547.
43. Sota A, Beck A, Wartenberg P, Gehl AL, Winter M, Wissenbach U, et al. TRPV6 channel function is involved in endometrial epithelial cell Ca2+ signaling and female mouse fecundity. Cell Mol Life Sci. 2025 Oct 7;82(1):346.
44. Suzuki Y, Kovacs CS, Takanaga H, Peng JB, Landowski CP, Hediger MA. Calcium channel TRPV6 is involved in murine maternal-fetal calcium transport. J Bone Miner Res. 2008 Aug;23(8):1249–56.
45. Bianco SD, Peng JB, Takanaga H, Suzuki Y, Crescenzi A, Kos CH, et al. Marked disturbance of calcium homeostasis in mice with targeted disruption of the Trpv6 calcium channel gene. J Bone Miner Res. 2007 Feb;22(2):274–85.
46. Fecher-Trost C, Lux F, Busch KM, Raza A, Winter M, Hielscher F, et al. Maternal Transient Receptor Potential Vanilloid 6 (Trpv6) Is Involved In Offspring Bone Development. J Bone Miner Res. 2019 Apr;34(4):699–710.
47. Sota A, Beck A, Wartenberg P, Gehl AL, Winter M, Wissenbach U, et al. TRPV6 channel function is involved in endometrial epithelial cell Ca2+ signaling and female mouse fecundity. Cell Mol Life Sci. 2025 Oct 7;82(1):346.