Abstract
Introduction: Alcohol or ethanol, as the correct chemical term, causes upon prolonged use in high amounts alcoholic liver disease (ALD), attributable to metabolic products and byproducts from enzymatic degradation of ethanol rather than to the relatively inert ethanol itself. The present review aimed to analyze mechanistic steps involved in ALD with a focus on molecular aspects related to the microsomal ethanol oxidizing system (MEOS), cytochrome P450 (CYP) 2E1, and ROS (reactive oxygen species). Apart from alcohol dehydrogenase (ADH), hepatic metabolism proceeds also via MEOS, which functions independently from ADH and catalase. MEOS is dependent upon various CYP isoforms with a preference of the CYP 2E1 isoform and requires not only NADPH + H+ as cofactor but also molecular oxygen. During alcohol metabolism via MEOS and CYP 2E1, and due to incomplete split of oxygen within the catalytic CYP 2E1 cycle, reactive oxygen species (ROS) are generated as toxic byproducts such as ethoxy radical CH3CH2O•, hydroxyethyl radical CH3C(•)HOH, acetyl radical CH3CHO•, single radical 1O2, superoxide radical HO•2, hydrogen peroxide H2O2, hydroxyl radical HO•, alkoxyl radical RO•, and peroxyl radical ROO•. Under mechanistic aspects, convincing evidence suggests that microsomal oxidative stress by radicals generated in liver microsomes through CYP 2E1 is directly involved in triggering early stages of ALD such as alcoholic fatty liver disease. Microsomal oxidative stress is also indirectly responsible for late stages of ALD such as liver cirrhosis, likely via modification of the immune system. In addition, radicals are generated by CYP 2E1 also in liver mitochondria causing mitochondrial oxidative stress and in the mucosa of the intestinal tract, suggesting some cotriggering pathogenetic role especially via endotoxins derived from the gut microbiome. In conclusion, MEOS, CYP 2E1, and ROS are primarily involved in triggering ALD via radical based toxic effects and alterations of the immune system.
Keywords
Alcoholic liver disease, Cytochrome P450 2E1, Endotoxins, Gut microbiome, Lipopolysaccharides, Microsomal ethanol oxidizing system, Microsomal oxidative stress, Mitochondrial oxidative stress, Reactive oxygen species
Introduction
Until the early sixties, the concept prevailed that alcoholic liver disease (ALD), also termed alcohol-related liver disease (ARLD), results from malnutrition commonly observed among individuals consuming chronically high amounts of alcohol rather than being causally related to the use of alcoholic beverages [1-3]. However, the malnutrition concept became a matter of debate because of the clinical observation that humans, even on a normal diet and without signs of underweight or malnutrition, were at risk of ALD. Under metabolic ward conditions and a nutritionally adequate diet, alcoholic fatty liver (AFL) developed, substantiating that short term use of alcohol combined with nutritionally adequate diets is responsible for the early manifestation of ALD [1]. These results obtained in humans were subsequently confirmed in experimental animal studies, whereby rats received alcohol in a nutritionally adequate diet [2]. Therefore and based on the pioneering work of Charles S. Lieber and his associates, the conclusion was reached that the alcoholic beverage itself rather than malnutrition causes the early stages of ALD [3], a proposal that became mainstream recognition [4-8]. However, alcohol is not a stable chemical when present in the liver but undergoes biotransformation to acetaldehyde as the first oxidation product [9-11]. These observations raised the question of whether metabolites of ethanol or metabolic byproducts such as toxic radicals generated during enzymatic degradation of ethanol rather than ethanol itself may be the causative agents in ALD.
Historically, hepatic cytosolic alcohol dehydrogenase (ADH) was considered as the principal enzyme responsible for alcohol metabolism, but this concept changed with the discovery of an additional pathway of enzymatic ethanol metabolism residing in the microsomal fraction of the hepatocytes corresponding to the endoplasmic reticulum [3]. This new enzyme was called the microsomal ethanol oxidizing system (MEOS) [12,13], leaving room for further characterization of its constituents [14-20].
In this article, prevailing conditions were analyzed, under which MEOS can contribute to the development of ALD. The focus was on hepatic reactive oxygen species (ROS) generated by the cytochrome P450 (CYP) with a preference for its CYP 2E1 isoform, one of the major constituents of MEOS. Analyzed were also similar conditions found for the gut microbiome. The terms alcohol and ethanol were used interchangeably.
Hepatic Alcohol Metabolism via MEOS
MEOS plays a significant role in alcohol metabolism [21-24]. Consensus exists that the liver is the primary organ for the metabolism of ethanol, with ADH and MEOS as the principal enzymes, whereas no significant role can be attributed to catalase [11,21-27] or nonoxidative metabolic pathways in humans [11]. Acetaldehyde in turn generated from ethanol via ADH and MEOS is further metabolized within the liver via the acetaldehyde dehydrogenase (ALDH) using NAD+ H+as reducing equivalents, which are identical for ADH but different from those for MEOS (Figure 1).
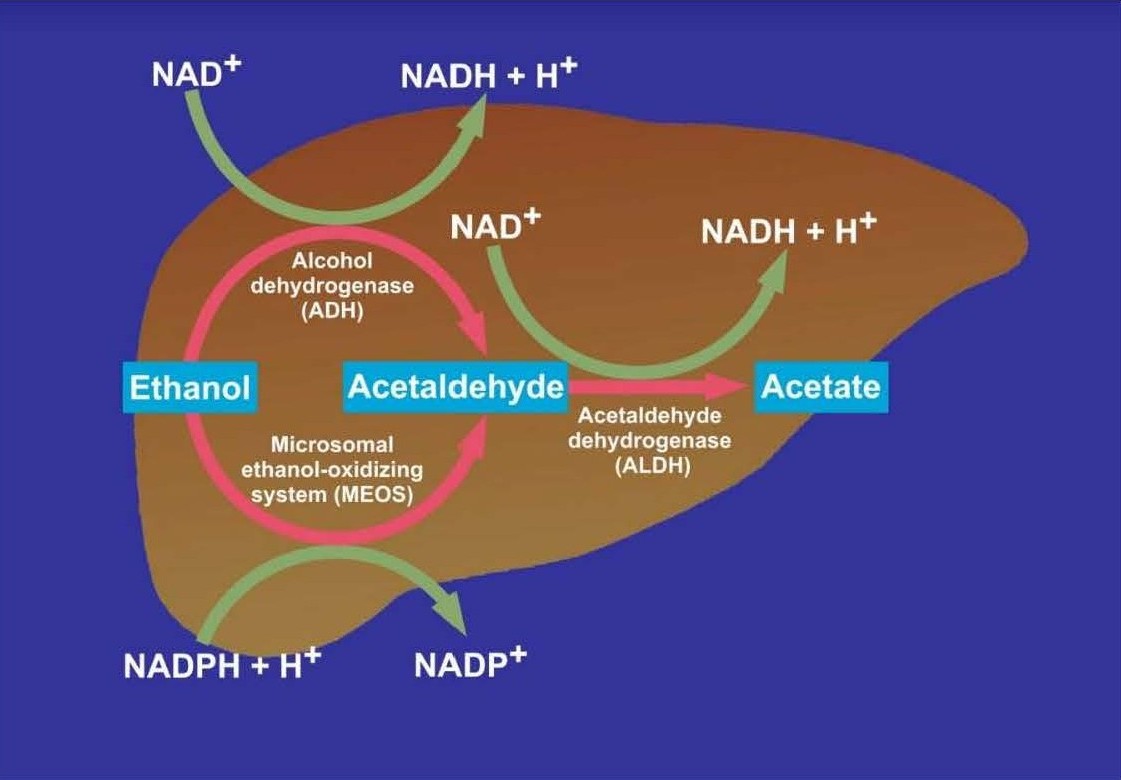
Figure 1. Major pathways of alcohol and acetaldehyde metabolism in the liver.
Whenever alcohol is present in the liver, both ADH and MEOS function synergisticly through an exchange of reducing equivalents [22]. Whereas alcohol metabolism via ADH produces reducing equivalents in form of reduced nicotinamide adenine dinucleotide NADH + H+ by reduction of NAD+, MEOS consumes reducing equivalents in form of reduced nicotinamide adenine dinucleotide phosphate NADPH +H+ with increasing demand due to enhanced MEOS activity under conditions of prolonged alcohol use and if alcohol concentrations are intermediate or high [22,23]. This synergistic process is facilitated by the nicotinamide nucleotide transhydrogenase (NNT), enabling the conversion of NADH + H+ into NADPH+ H+ [11,28]. Reducing equivalents in form of NADH + H+ are also consumed by the cytochrome b5 in connection with the NADH dependent cytochrome b5 reductase, both are constituents of the hepatic microsomal membranes [17,29,30]. Although several sources are known, which provide or consume reducing equivalents, the best studied is the respective interconnected action of ADH and MEOS (Figure 2).
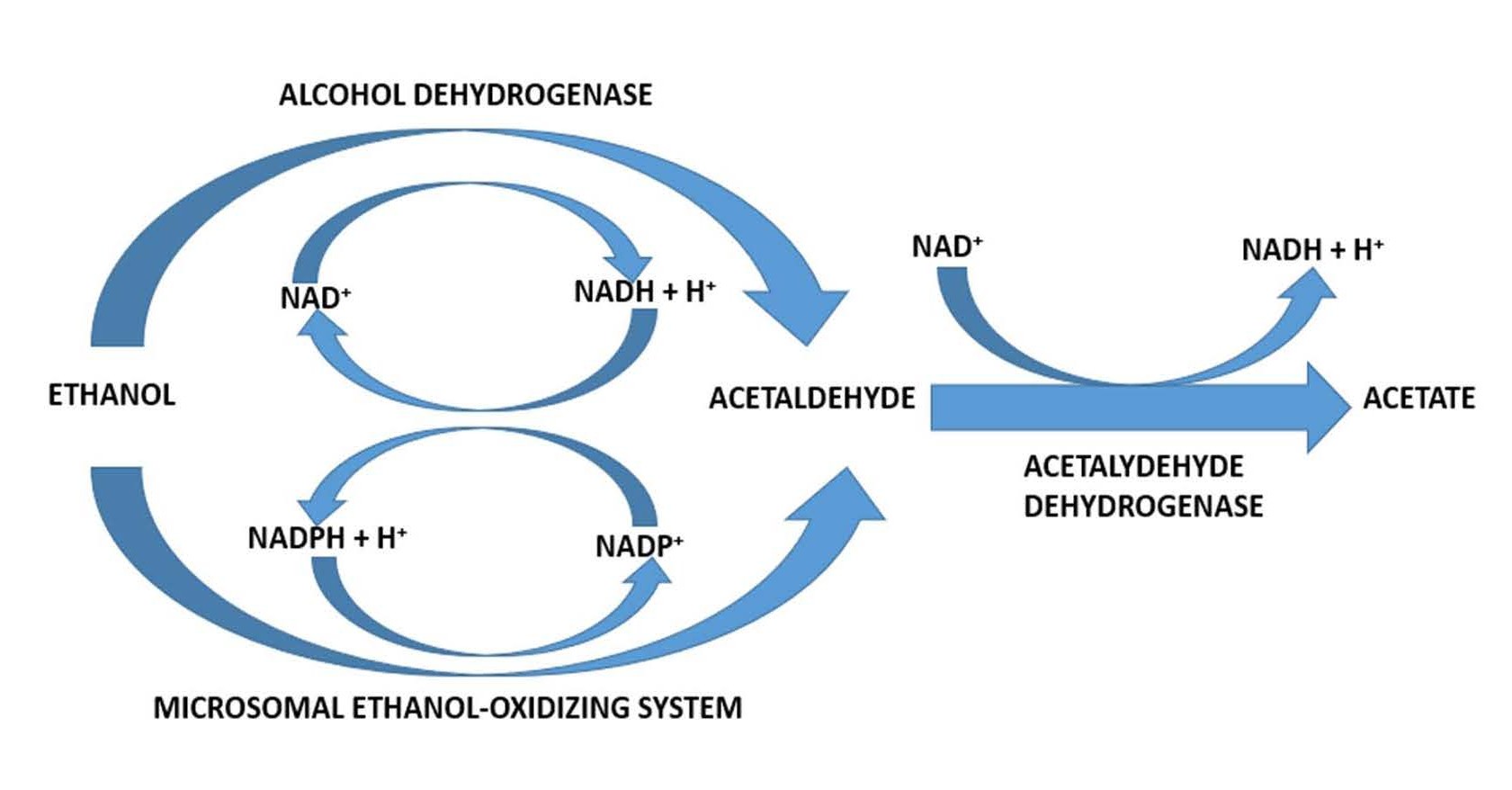
Figure 2. Interconnected action of hepatic alcohol dehydrogenase (ADH) and the microsomal ethanol oxidizing system (MEOS). ADH produces reducing equivalents, which are used by MEOS, showing that both enzymes depend on each other. The original figure was published in an earlier report [22].
There is good evidence that the increased metabolic rate of alcohol observed following prolonged alcohol consumption is attributable to the increase of hepatic MEOS activity [21-24] rather than to ADH activity, which remained unchanged following chronic alcohol use [11,21-24]. The interaction of alcohol with the hepatic endoplasmic reticulum is also supported by the proliferation of the smooth endoplasmic reticulum following prolonged alcohol consumption, established upon electron microscopy in humans [31] and animals [32]. Harvested in the microsomal fraction after homogenization and ultracentrifugation of the hepatocyte, the membranes of the smooth endoplasmic reticulum exbibit increased activities not only of MEOS by 44% but also of the microsomal NADPH cytochrome c reductase by 43%, associated with enhanced contents of microsomal CYP by 57% and microsomal phospholipids by 26% [15]. Concomitantly, the activities of hepatic microsomal drug metabolizing enzymes such as the demethylases for aminopyrine and ethylmorphine were significantly increased following prolonged alcohol use, with higher activities in the smooth microsomes than in the rough microsomes [33].
Nature and Constituents of MEOS
Differentiation of MEOS from ADH and catalase
It was early described that some microsomal preparations containing MEOS activity may be contaminated by ADH and/or catalase activities to the various extent from batch to batch and depending on specific preparation conditions [13]. Therefore, studies have been carried out using inhibitors of ADH and catalase that showed preservation of MEOS activity despite complete inhibition of ADH and catalase [13,14]. Later studies also focused on issues of substrate specificities because catalase peroxidizes only alcohols with short aliphatic chains but not with long ones. For instance, pentanol, butanol, and propanol are perfect substrates for the microsomal system but not for catalase [16,18,29].
Solubilization and column chromatography of MEOS
Column chromatographic studies based on ion exchange and a linear potassium chloride (KCl) gradient for the elution procedure have previously shown in 1972 and for the first time that MEOS retained its activity after being physically separated from ADH and catalase, characterizing its independency from ADH and catalase [15]. In retrospect, at that time it was fascinating to see that constituents of the microsomal membranes preserved MEOS activity despite the disruption of the membranes by ultrasonication and the use of the detergent sodium deoxycholate. In 1978 and using a different approach, the direct oxidation of ethanol by a reconstituted system free of catalase and alcohol dehydrogenase confirmed that MEOS functions without ADH and catalase [34], in support of the earlier report using ion exchange column chromatography with a linear KCl gradient [15] and of similar studies that used a stepwise KCl gradient to speed up the elution procedure [17,18,29]. The separation of MEOS from ADH and catalase activities published in 1972 [15] was independently confirmed in 1982 [35]. These two studies [34,35] settled a long standing controversy around the nature of MEOS [30], attributed by some investigators erroneously to ADH and/or catalase as discussed before [15-24] and now updated [30]. The elution pattern of the solubilized and isolated MEOS revealed the highest activity in elution fractions that contained CYP together with the reductase and phospholipids, with missing MEOS activity when only the reductase was present with or without phospholipids, or if phospholipids alone were present (Figure 3) [15]. However, MEOS activity did not parallel the CYP content, suggesting differences of turnover numbers in MEOS activity over CYP content in line with different CYP isoforms characterized by variable affinities for ethanol.
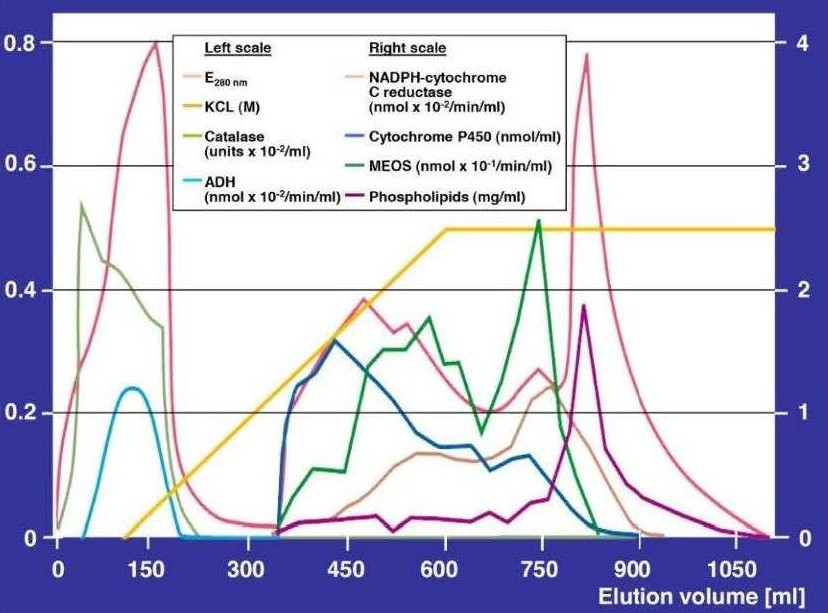
Figure 3. Elution pattern with separation of the microsomal ethanol oxidizing system (MEOS) from catalase and alcohol dehydrogenase (ADH) activities.
Using column chromatography, eluates started with the void volume recovered up to around 220 mL. Here, the highest peak represents the protein curve assessed as E280 nm, and the peak below that is the catalase peak, whereas ADH shows up as the lowest peak. Starting with an elution volume of around 330 mL, then microsomal components begin to appear. The first peak represents cytochrome P450, the second peak represents E280 nm, followed by a third peak with two shoulders and by a fourth peak representing MEOS. At around 770 mL, the reductase peak emerges, followed by the phospholipid peak at around 790 mL elution volume. Modified from the original figure published in a previous report [15].
With CYP, NADPH CYP reductase, and microsomal phospholipids, MEOS shares constituents similar to other hepatic microsomal drug metabolizing enyzymes [15]. These three components are required for the function of MEOS, as outlined and summarized in Methods of Enzymology [14] and shown by reconstitution experiments [19,20,34], confirming previous studies using column chromatography (Figure 3) [15,17,18,29]. Whereas phospholipids are in support of the enzymatic process [15,19,20,34], interactions of CYP and the reductase can now be shown for MEOS oxidizing ethanol to acetaldehyde (Figure 4).
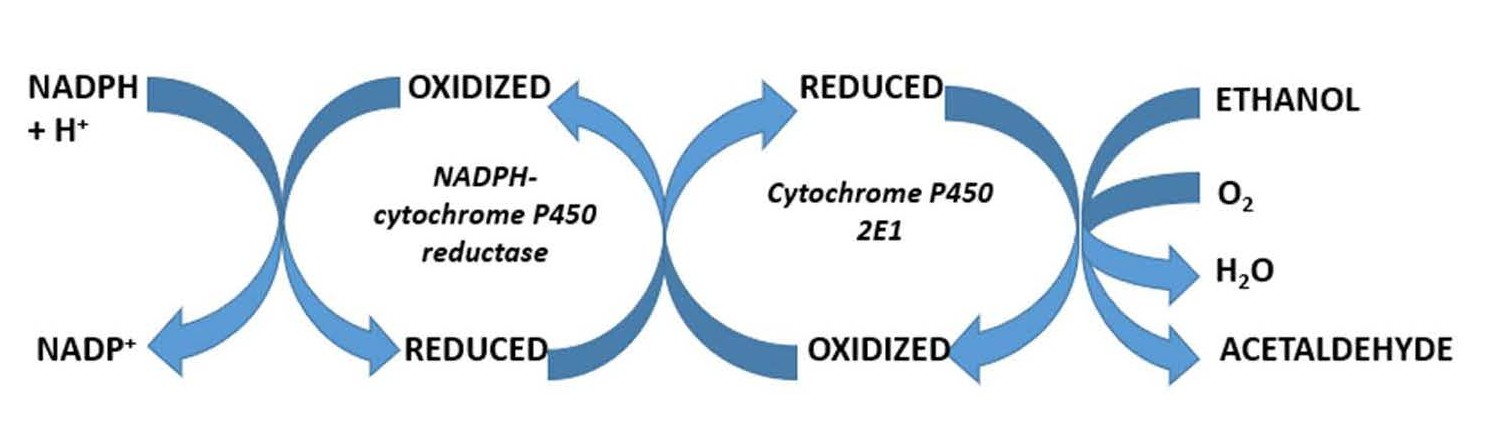
Figure 4. Key microsomal components of MEOS, whereby phospholipids likely interact between CYP 2E1 and the NADPH CYP reductase.
Definition of MEOS
Final definition
Using preferentially the characteristic features following its chromatographic isolation [15,17,18,35] and its reconstitution from microsomal constituents [19,20,34], MEOS is now finally well defined [22-24,30,34]: (1) MEOS converts ethanol to acetaldehyde in the absence of ADH and catalase [15,17,18,29,34,35]; (2) MEOS exerts it maximum activity at a physiological pH of 6.9 - 7.5 [17]; (3) MEOS has a Michael-Menten constant of 7.2 mM for ethanol and is thereby active at intermediate and higher alcohol concentrations found among individuals consuming alcoholic beverages in modest and higher amounts [17]; (4) near maximum rates of ethanol oxidizing activity were measured with ethanol concentrations of 30 mM and above [17]; (5) the apparent Michael-Menten constant of the purified MEOS for oxygen was 8.3 μM, whereas near maximum ethanol oxidizing activity was observed with oxygen concentrations of 30 μM and above [17]; (6) MEOS activity increases after chronic alcohol consumption and accelerates thereby ethanol removal from the body [12-15,21]; (7) as opposed to CYP 2E1 of MEOS residing in the microsomal membranes [15,17,20], hepatic mitochondrial CYP 2E1, which employs adrenodoxin to transfer electrons to CYP [36-41], is induced by ethanol intoxication [41] but has likely no significant role in the overall alcohol metabolism [36], and is by no means part of real MEOS [30]; (8) instead, MEOS is promoted by the microsomal constituents CYP with its various human isoforms CYP 1A1, CYP 1A2, CYP 2A6, CYP 2B6, CYP 2D6, CYP 3A4, including CYP 2E1 as its most active isoform [42], the NADPH CYP reductase [15,19,20,34], and phospholipids [15,19,20,34], serving likely in form of lipid peroxides as a platform for the CYPs and the NADPH CYP reductase to integrate into and thus allow for a better electron flow from the reductase to reduce the ferric CYP to the ferrous state followed by binding to oxygen [30]; (9) the involvement of microsomal components like phospholipids, the NADPH CYP reductase, and various CYP isoforms makes MEOS to a microsomal multi-CYP isoform system [30]. Thus, the term MEOS characterizes total microsomal ethanol oxidation, achieved through the microsomal CYP catalytic cycle with ethanol as the substrate (Figure 5).
Mechanistic actions
The mechanistic steps involved in MEOS activity have been studied and discussed in detail [3,14-20,23,24,29,30,34-36,43-50] and may tentatively be summarized: (1) MEOS activity as isolated during column chromatography is insentitive to superoxide dismutase, disregarding superoxide radicals as sole intermediates in ethanol metabolism via MEOS [17]; (2) likewise, H2O2 itself is incapable of promoting ethanol oxidation via the isolated MEOS eluted during column chromatography in fractions containing no catalase but CYPs [17]; (3) although neither superoxide radicals nor H2O2 alone can promote microsomal oxidation, under physiological conditions they may react together to slowly yielding hydoxyl radicals required for MEOS [30]; (4) MEOS requires for its activity reducing equivalents in form of NADPH + H+ as cofactor and molecular oxygen [17]; (5) the combined action of NADPH + H+ and molecular oxygen within the microsomal CYP catalytic cycle (Figure 5) leads via incomplete split of molecular oxygen to the generation of ROS, partially used by the radical scavenging ethanol in order to be oxidized and to produce acetaldehyde [30]; and (6) ROS not used for microsomal oxidation of ethanol can trigger liver injury [30].
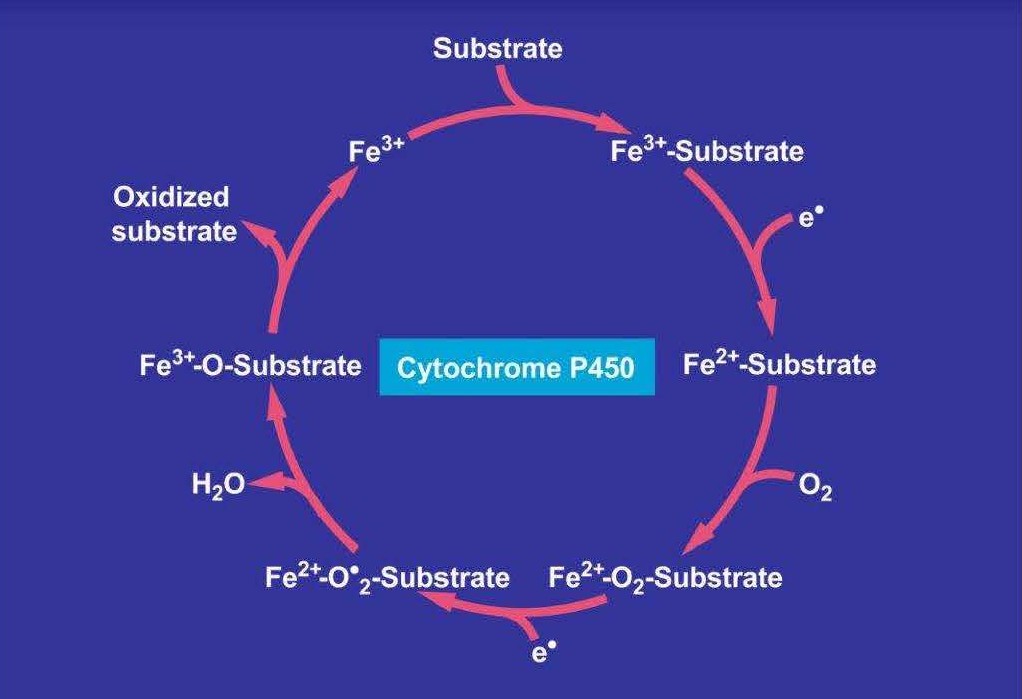
Figure 5. Catalytic CYP 2E1 cycle of MEOS, oxidizing ethanol as substrate and providing acetaldehyde as oxidized substrate.
Liver Injury by Acetaldehyde, CYP 2E1, and Reactive Oxygen Species
Vicious circle of acetaldehyde
In humans consuming high amounts of alcohol over a prolonged time, the hypothesis of a vicious circle of acetaldehyde in the liver has been proposed [9]. This hypothesis is based on the following conditions and steps: (1) acetaldehyde is generated during ethanol metabolism via MEOS, which is increased in activity following prolonged alcohol use and contributes to the removal of alcohol from the body [30]; (2) as a result and as expected, the liver is confronted with high amounts of acetaldehyde, commonly viewed as hepatotoxic [9-11]; (3) increased acetaldehyde levels in the liver, in turn, impair mitochondrial functions associated with a reduction of the mitochondrial ALDH activity [9,10]; (4) the decreased acetaldehyde metabolism increases the acetaldehyde level in the liver, which further impairs mitochondrial functions including ALDH activity [9]; (5), thus, a vicious circle develops, starting with increased acetaldehyde levels in the liver, followed by mitochondrial impairment and again increased hepatic acetaldehyde levels [9-11]. The original vicious circle published in 1975 [9] should be updated by aspects of ROS formed in the course of microsomal oxidation via CYP 2E1 and other CYP isoforms [30,43-54], contributing to mitochondrial damage and the generation of toxic ethoxy radicals from acetaldehyde [30] and completing the vicious circle [9].
The action of acetaldehyde is not limited to the liver. Part of the acetaldehyde leaves the hepatocytes, enters the circulatory system, and is detected in the blood with higher levels in individuals with an alcohol problem compared to controls without this problem [55]. Consequently, acetaldehyde can reach via the bloodstream virtually all cells of any organ including the brain, where it may contribute following condensation reactions with serotonin to trigger the mechanism of alcohol dependence, a fascinating but still controversial aspect (Figure 6) [56].
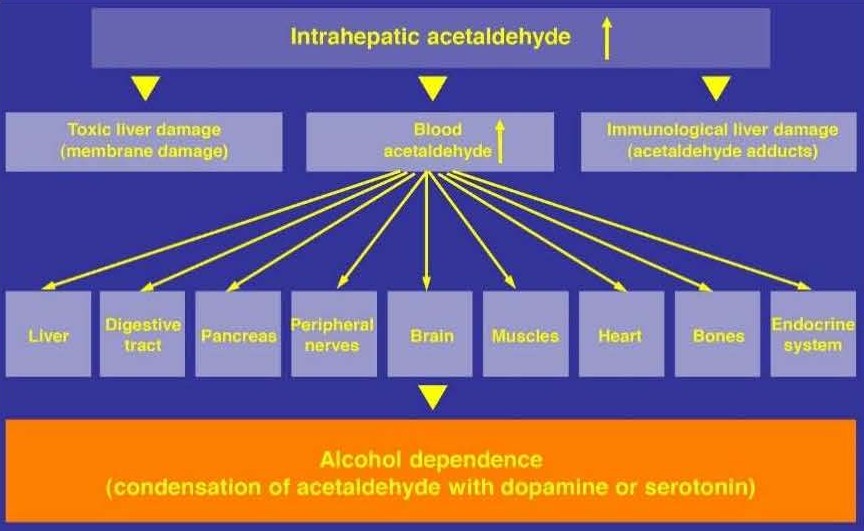
Figure 6. Actions of acetaldehyde.
The increasingly generated acetaldehyde in the liver spills over in the blood and reaches many organs, which are injured by direct toxic attacks or through condensation products. Alcohol dependence is considered to be triggered by the condensation of acetaldehyde with dopamine or serotonin. Symbol ↑: Increase.
CYP 2E1 and reactive oxygen species
Alcohol and the microsomal CYP catalytic cycle: The metabolism of ethanol via CYPs located within the microsomal membrane of the hepatocyte proceeds through the CYP catalytic cycle as follows in short (Figure 5) [30,54]: (1) Substrates like ethanol bind in proximity to the heme group of the oxidized ferric CYP [30,54]; (2) substrate binding to the heme of CYP leads to displacement of water at the sixth ligand to the heme iron, changing the spin state of the iron from low spin to high spin [54]; (3) substrate binding also induces the transfer of the first electron from NADPH via CYP reductase to reduce the heme from the ferric state (Fe3+) to the ferrous state (Fe2+) [54]; (4) molecular oxygen binds to the resulting substrate-ferrous CYP [54]; (5) the second electron may then come from either the NADPH dependent CYP reductase or may be provided by reduced cytochrome b5 via the NADH cytochrome b5 reductase, thereby reducing the Fe2+-O2 adduct to give a short-lived, highly reactive intermediate complex [54]; (6) upon decomposition of this intermediate complex, one molecule of water is released, highly reactive species are formed, and the substrate like ethanol is oxidized to acetaldehyde [30,54]; (7) after the product has been released from the active site, the enzyme returns to its original state, with a water molecule returning to occupy the distal coordination position of the iron nucleus [54]; (8) under mechanistic aspects, there is no evidence that superoxide radicals are the triggering compounds involved in MEOS activity, because superoxide dismutase as a potent scavenger of superoxide radicals fails to reduce the rate of ethanol metabolism in the isolated MEOS [19], findings confirmed for the reconstituted system [57]; (9) instead, based on the inhibitory effect of the hydroxyl radical scavenger sodium formate on MEOS activity of the isolated MEOS fraction as published in 1974 [17] and confirmed in whole microsomes as reported in 1975 [19], hydroxyl radicals were assumed as the most likely candidates participating in microsomal ethanol oxidation, especially also because ethanol itself is known for its hydroxyl radical scavenging potency [19]; and finally (10), subsequent studies published since 1982 discussed and confirmed the role of hydroxyl radicals in microsomal ethanol oxidation [47,49,50,52,53,57-59], substantiated also by other inhibitory studies using potent hydroxyl radical scavengers such as dimethylsulfoxide, benzoate, mannitol, and thiourea [58].
There were considerations that the microsomal oxidation may not involve a specific CYP dependent mechanism but rather results from hydroxyl radical formed primarily in iron-catalyzed Haber-Weiss and Fenton reactions [53]. However, the activity of MEOS recovered in eluates during column chromatography activity was always analyzed using incubation media that were supplied by dinatrium- EDTA, known for its forming stable complexes with Fe3+and removing thereby soluble Fe3+ [15,17-19,30]. This rules out any contribution of iron of nonenzymatic origin, possibly present in the incubation medium, and clearly defines MEOS as an enzymatic system rather than an artifact of a nonenzymatic, artificial system [30]. Support for MEOS as a real enzymatic system is also provided by its inactivation following boiling and the requirement of molecular oxygen [16,20]. Although nonenzymatic iron cannot replace constituents of MEOS, it certainly helps enhance microsomal hydroxyl radical formation if the iron is present in sufficient amounts in the liver [53,60], which plays a fundamental role in heme catabolism and iron recycling [60]. However, under ordinary real world conditions outside of experimental in vitro studies, increased hepatic iron contents have not been reported in healthy individuals but are hallmarks of genetic hemochromatosis, nonalcoholic fatty liver disease, and hepatitis C virus infection [60], including ALD [61].
Surprisingly, microsomal ethanol oxidation may proceed via two different pathways in a reconstituted microsomal system consisting of CYP and the CYP reductase purified from rats treated by phenobarbital [59]. According to this proposal, one of these pathways involves the purified NADPH dependent CYP reductase itself, which oxidizes ethanol in the absence of CYP and is inhibited by superoxide dismutase [59]. These data and conclusions are different from those of other studies showing lack of ethanol oxidation by the NADPH CYP reductase at the end of the chromatographic elution (Figure 3) [15], and lack of ethanol oxidation was found in the reconstituted system with purified NADPH CYP reductase [20]. The second pathway consists of CYP and CYP reductase as the major components of MEOS activity and is inhibited by superoxide dismutase, which would imply superoxide radicals playing a role in MEOS activity, and competing hydroxyl radical scavengers, suggesting a role of hydroxyl radicals [59]. However, earlier studies found no inhibition of MEOS by superoxide dismutase [17,20,58]. Overall agreement exists that hydroxyl radicals play a major role in MEOS activity [18,20,47,58,59].
ROS and oxidative stress : Within the hepatocyte, ROS can be generated in non-organelles via NADPH oxidase, xanthine oxidase, cyclooxygenases, and lipooxygenases, but are preferentially generated in two different subcellular compartments, the endoplasmic reticulum and mitochondria [30]. The endoplasmic reticulum corresponds to the microsomes [18,20,47,58-60], a process that may cause adverse reactions including injury collectively known under the term “microsomal oxidative stress” [60]. ROS generation in the mitochondria [37-41,60] leads to injury under the term of mitochondrial oxidative stress [60]. Lipid peroxides are the assumed primary intermediates eliciting microsomal and mitochondrial injury through oxidative stress [60] but other radical species commonly generated may contribute [7,23]: ethoxy radical CH3CH2O•, hydroxyethyl radical CH3C(•)HOH, acetyl radical CH3CHO•, single radical 1O2, superoxide radical HO•2, hydrogen peroxide H2O2, hydroxyl radical HO•, alkoxyl radical RO•, and peroxyl radical ROO•. Currently, the quantitative role of microsomal CYP 2E1 versus mitochondrial CYP 2E1 in ALD initiation and progress has not yet firmly been settled. Through various processes including signaling mediators and changes of immune systems, ROS may directly be involved by triggering early stages of ALD such as AFL or indirectly via immune mechanisms causing late stages of ALD like cirrhosis (Figure 7) [22,23].
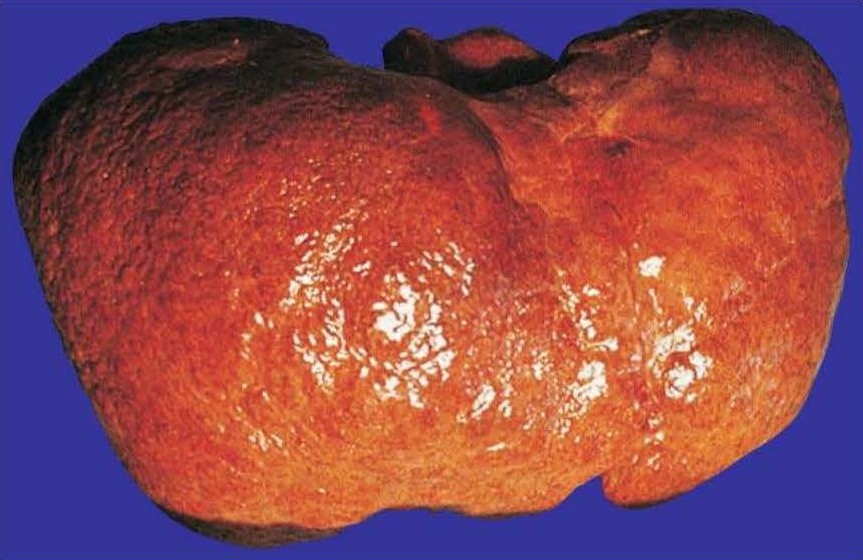
Figure 7. Macroscopic picture of alcoholic cirrhosis. The granular surface of the liver reflects the regenerative nodules, which can be seen upon histological evaluation.
Immune systems, ROS, and the Gut Microbiome
Liver innate and adaptive immune systems
Special immune systems modify the natural course of various liver diseases including those caused by toxins such as alcohol [22,23,62-66] and drugs [67-70]. To become functional, the quiescent innate immune system must be activated to the adaptive immune system [67-70]. The activation allows the liver for its adaptation to various immune challenges. They may relate, for instance, to degradation products of ingested toxins such as reactive metabolites of alcohol formed as byproducts in response to the high oxidative state of the liver during ethanol metabolism [62]. Alternatively, they may relate to bacteria [71,72] in the context of the gut microbiome with their endotoxins as inducers of proinflammatory T helper cells and regulatory T cells in the intestinal tract [8], conditions of relevance for patients with ALD [22,23,30].
Compelling evidence exists that an immune component may be involved in the development and progression of ALD [8,62-66], although non-immune mechanisms are also to be considered at least in some patients with acute alcoholic hepatitis (AH), who do not respond to treatment by cortisol [73]. Support for the immune component is provided by the detection of circulating autoantibodies, infiltration of immune cells in the liver, and the detection of hepatic aldehyde modified proteins in patients with ALD [62]. The immune issue in ALD is known for three decades and is exhaustively considered in many publications including some selected ones [8,27,22,23,30,62-66]. As expected, a broad range of different mechanistic steps have been proposed and variably covered, open for discussion [8,62-66]. Under clinical aspects, immune challenges in ALD with a focus first on ROS and then the gut microbiome will be considered as the most important areas of interest.
Immune systems and ROS
It is generally accepted that AFL as the first stage of ALD is primarily caused by redox changes due to increased production of NADH + H+ and disturbances in fatty acid metabolism rather than related to immune systems [3,27]. Instead, immune mechanisms prevail in later stages such as alcoholic steatohepatitis (ASH), AH, or alcoholic cirrhosis (AC) with or without hepatocellular carcinoma (HCC), although the exact mechanistic steps, by which alcohol or its byproducts initiate adaptive immune responses, are not entirely understood [63-65]. This is unexpected because of a large number of published studies on complex interactions between ethanol, its metabolic byproducts, and the immune systems [3,22,23,27,62-66]. Acetaldehyde and various toxic radicals, derived from actions of CYP 2E1 and included in the broad term of ROS, are considered as causatives in patients with progressing to late stages of ALD following modification of immune systems [22,23,27,62-66].
First, toxic is both, acetaldehyde itself with its normal chemical structure [7,9,11,27] or as radical structure generated during the microsomal oxidation [62,63,64,66], although respective radicals likely will have a higher power to initiate immune modification in line with microsomal oxidative stress. In addition, mitochondrial oxidative stress may contribute if ethanol is metabolized by mitochondrial CYP 2E1 [60]. As a result, immunogenic acetaldehyde (AA) protein adducts are produced and found in individuals with an alcohol problem, one of these adducts has been called malondialdehyde-acetaldehyde adduct (MAA) [62,63]. Clinical and experimental studies have shown that MAA levels are increased in the serum, correlated with the severity of human ALD [62], and produced experimentally strong antibodies to the adduct, substantiating that MAA initiates the immune system to respond [62,63]. These responses were achieved by upregulating adhesion molecules, inducing proinflammatory cytokines, producing T-cell responses, and increasing profibrotic response [62].
Second, all other forms of ROS such as ethoxy radical, hydroxyethyl radical, acetyl radical, singlet radical, superoxide radical, hydrogen peroxide, hydroxyl radical, alkoxyl radical, and peroxyl radical may be formed during NADPH dependent microsomal ethanol oxidation and are likely responsible for triggering ALD through immune modifications related to microsomal oxidative stress that would reflect the action of microsomal CYP 2E1 [23,24,27,30].
Microsomal and mitochondrial oxidative stress imply that toxic intermediates and radicals are primarily injurious to microsomes and mitochondria, with changes of integral constituents of organelle membranes, destroying thereby the membrane integrity and reducing the functions of the membranous enzymes [22,23,30,37-41,60,62]. Membrane constituents of liver cell organelles including microsomal and mitochondrial ones have been described in detail, with a focus not only on proteins in form of enzymes but also on cholesterol and phospholipids [74-76]. In terms of chemical structure, phospholipids are commonly made up of a phosphate group head and one or two fatty acid tails, all connected with a third molecule, glycerol. Fatty acids of phospholipids contain either saturated, monounsaturated or polyunsaturated chains, whereby phospholipids with higher unsaturated fatty acids are more likely targets to lipid peroxidation triggered by radical metabolites. In the peroxidized formation, phospholipids will not only help get MEOS running [23,30] but also initiate injury in microsomal and mitochondrial membranes by attacking proteins. This is evidenced by the observation of immune or auto-immune based parameters such as circulating lymphocytes specific to hepatic antigens [62], circulating antibodies specific to metabolite adducts involving hydroxyl radicals interacting with proteins [62,63], and circulating anti-phospholipid antibodies, which are found in up to 80% of patients with alcoholic hepatitis or cirrhosis [63]. More specifically, lipid peroxidation markers are increased in both the liver and the serum of patients with ALD, with elevated circulating IgG towards protein adducted byproducts derived from lipid peroxides [63]. In line with these findings, histology reveals that the liver infiltrates rich in neutrophils and characteristic of alcoholic hepatitis also contain both CD8+and CD4+ T-lymphocytes, which may produce cytokines.
Immune systems and gut microbiome
The gut microbiome has attracted much interest as a contributing factor in ALD [7,8,62-66]. Following initial proposals first published in 1984 by Christian Bode and his group from Germany that a gut microbiome exists relevant in patients with ALD [77-83], credit to this discovery was rarely given during the subsequent years although consensus now exists that endotoxins provided by the gut microbiome may initiate or promote disease progression [8,22,27,84-90] including HCC [90]. Since alcohol metabolizing enzymes such as MEOS with CYP 2E1 are present in the mucosa of the gastrointestinal tract of animals and humans [91-93] including CYP 2C, 2E1, 3A4, and 3A5 in the mucosa of the human colon [93], the discussion expanded on the relation of MEOS, CYP 2E1, the gut microbiome, and ALD [94-96]. Good evidence exists that intestinal CYP 2E1 is a mediator of the alcohol dependent gut leakiness [95], whereby intestinal CYP 2E1 seems critical under binge alcohol conditions and potentiates the gut leakiness [94]. Of additional importance is the observation that MEOS activity is found in the intestinal tract [91,92], signifying that intestinal CYP 2E1 is active and toxic acetaldehyde is produced by mechanisms involving CYP, the NADPH reductase, and phospholipids in a similar way as in liver microsomes whereby injurious ROS are generated [22,23,30,96].
According to current knowledge, the link of the gut microbiome with ALD may tentatively be described with the following steps: (1) under normal conditions, the circadian clock regulates the physiology and function of the intestinal tract but alcohol causes circadian disorganization [97], (2) alcohol consumption upregulates mucosal CYP 2E1 content and MEOS activity in the intestine leading to the generation of acetaldehyde and ROS [91-94,96], (3) alcohol also modulates the mucosal immune system of the gut [82] with dysregulation of the innate immunity [94], (4) alcohol has direct injurious effects on the mucosa of the intestinal tract [82]; (5) consumption of alcohol modifies the composition of the intestinal microbiotica [98] and facilitates bacterial overgrowth in the small intestine, leading to the production of endotoxins, which represent chemically lipopolysaccharides [97,98] derived from the outer membrane of gram negative bacteria of the intestinal tract [92,97], (6) alcohol leads to functional and morphological changes of the gut mucosa, disrupts the intestinal barrier function [94], impairs the mucosal integrity [82], and (7) promotes gut leakiness, possibly through a mechanism involving the tight junction protein occludin [97], (8) the leaky gut allows for the transition of larger chemicals like endotoxins and other bacterial toxins from the gut lumen to the portal vein [98], (9) endotoxins enter the liver and the systemic circulation, where they can be quantitatively assessed causing endotoxemia [98], and finally (10), endotoxins in the liver may contribute to the initiation or progress of ALD [98]. A first insult may prime the liver to another hit by endotoxins because patients with other diseases or conditions unrelated to alcohol like irritable bowel syndrome, celiac disease, food allergies, cancer, and cardiovascular disease may also have increased endotoxin levels but no associated liver disease [98]. Despite many studies [62-66,77-83,98], the quantitative role of endotoxins as causes of ALD remains to be established, although data in humans are limited [98].
Overview of Mechanistic Steps in ALD
In addition to endotoxins derived from the gut microbiome [8,62-66,86-90], both hepatocytes and nonparenchymal cells of the liver are involved in the development of ALD [22,23], with hepatocytes governing the metabolism of ethanol and acetaldehyde [22]. To maintain the normal biologic and immunologic functions and regulate the homeostasis of the liver, the various cells are closely connected through signaling mediators. For a quick overview, the pathogenetic complexity of ALD is illustrated as an example for alcoholic hepatitis and includes issues of hepatocytes and several nonparenchymal cells of the liver like Kupffer cells, stellate cells, and sinusoidal cells, the role of mediators as signaling pathways like various growth factors, the tumor necrosis factor, interleukins, and interferon, considering also the generation of multiple ROS (Figure 8) [22]. Under discussion is also the genetic polymorphism of CYP 2E1 as a risk factor of ALD [99].
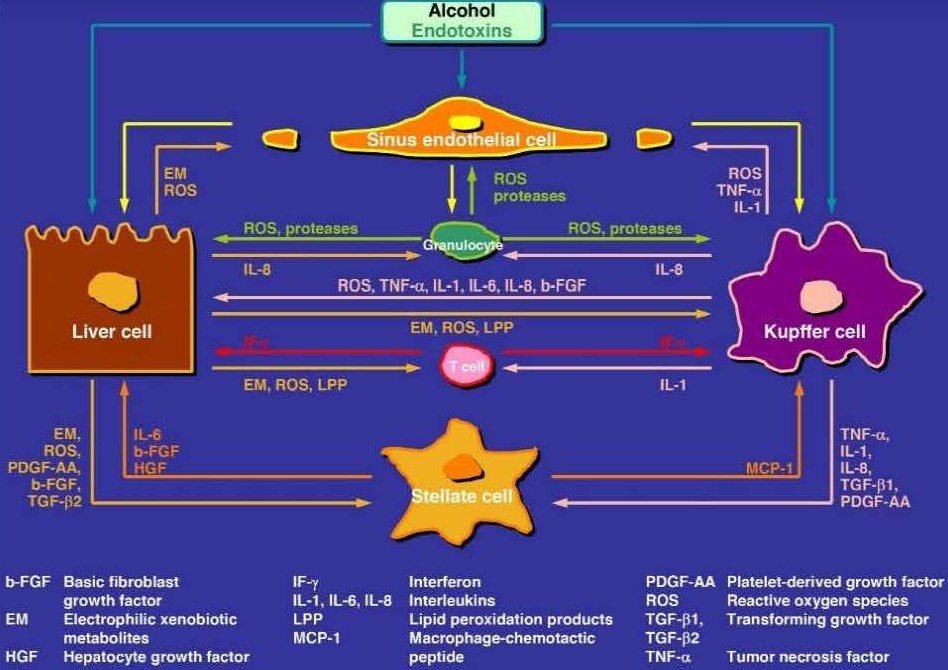
Figure 8. Hypothetical steps leading to alcoholic hepatitis (AH). The pathogenesis of alcoholic hepatitis involves various mediators and cell types (hepatocytes, Kupffer and stellate cells) of the liver, some of the steps need confirmation and are therefore hypothetical. The possible interactions between these cells and mediators are overwhelming and mostly derived from individual experimental studies. Interactions may be stimulatory or inhibitory in the context of the inflammatory state, but are difficult to be foreseen in a patient with AH. The figure was published in a recent report [22].
Clinical manifestation of ALD is variable and presents as five stages: AFL, ASH, AH, AC, and alcoholic HCC [22]. Although some overlap is possible in terms of liver histology, there is not a single hit that could explain all five stages of the ALD disease. Consequently, a hypothesis was proposed that classified ALD as a multihit disease [22]. The concept of a multihit disease as outlined for ALD is under similar discussion also for a variety of other diseases including cancer, chronic disabling diseases, and more recently, obesity with nonalcoholic fatty liver disease (NAFLD) or nonalcoholic steatohepatitis (NASH) [22]. The proposed five-hit working hypothesis of ALD is based on highly complex clinical as well as experimental conditions and may help shedding more light on the mechanistic steps leading to ALD. In more detail, the first hit depends on ADH and occurs through the generation of NADH + H+, which leads to an increased NADH + H+/ NAD+ ratio, stimulates hepatic fatty acid synthesis, and increases α-glycerophosphate-trapping fatty acids [22]. In addition, acetaldehyde is generated, which impairs hepatic mitochondrial functions including fatty acid oxidation [22]. This first hit explains at least in part the development of AFL. The second hit is described as the transition from AFL to ASH, most likely triggered by the increased production of acetaldehyde via MEOS and of ROS with its capacity for irreversible covalently binding to cellular macromolecules, including membrane proteins and phospholipids [22]. The third hit initiates a more severe liver injury stage, whereby ASH is the precursor in most, but certainly not all patients with AH. Steatosis is not any more a characteristic feature but is now replaced by necrosis, apoptosis, and inflammation. At this stage, the injury becomes more severe and presents with increased fibrosis and as a self-perpetuating process. Immunity aspects gain additional relevance, because alcohol modifies the innate and adapted immune system, which may explain the individual differences of susceptibility for ALD [22]. With the third hit, the disease may approach a point of no return [22]. The fourth hit is dominated by increased fibrosis resulting from increased collagen formation. This allows for a clinically unrecognizable transition from AH with fibrosis to irreversible cirrhosis. However, cirrhosis can also develop without ASH or AH [22]. In rare cases, a fifth hit initiates the development of HCC, mostly occurring in patients with cirrhosis. This final hit scenario of carcinogenesis is triggered by acetaldehyde and ROS through the generation of DNA adducts, which promote mutagenesis, and interference with methylation, synthesis, and repair of DNA. These overall events will enhance the carcinoma susceptibility, keeping in mind that ethanol itself is not a carcinogenic chemical [22].
Experimental studies provided additional support for the potential role of CYP2E1 and ROS in ALD [100-103]. In CYP2E1 over-expressing mice, ROS production was increased and ALD severity enhanced [100], while in CYP2E1 knock-out animals, ALD was less severe [101]. When clomethiazole (CMZ), a strong CYP2E1 inhibitor [102], was given to rats with ALD, the disease significantly improved [103], suggesting that CYP2E1 activity advances ALD. Consequently, additional research approaches are to be expected in search for more inhibitors of CYP 2E1 as treatment modalities, although admittedly, achieving alcohol abstinence should be the primary choice [73]. If alternatives are needed, phytochemicals, or other naturebased chemicals could be helpful [104].
Apart from mechanistic and therapeutic aspects, MEOS, CYP 2E1, and ROS are seemingly implicated in ALD through uncovering potential diagnostic biomarkers [105-108]. For instance, there were exciting studies on circulating blood exosomes [105,106]. In patients with a history of prolonged alcohol use and animals exposed to binge alcohol or repeated doses, extracellular vesicles were detected in the blood containing CYP isoforms, namely CYP 2E1, 2A6, 1A/2, and 4B in patients, and CYP 2E1, 2A3, 1A/2, and 4B in animals [105]. However, their utility as clinical diagnostic biomarkers has still to be determined [105,106]. Similarly, serum activities of glutamate dehydrogenase (GDH) may be a good diagnostic marker for alcoholism or early stages of ALD like AFL, with a 5-fold increase in the serum of patients with AFL, which was significantly different from controls and showed little variability [107,108]. GDH is a mitochondrial enzyme originating from the centrilobular region of the liver, which is rich in CYP 2E1 and thereby MEOS [109]. Whether blood determination of endotoxins, triggered by intestinal CYP 2E1, is a useful diagnostic tool in a clinical setting remains to be established [77-90].
Apart from CYP 2E1 [22-24,100-103], new experimental data suggest that nonCYP2E1 isoforms such as CYP 2A5 or CYP 2A6 may interact with alcohol and CYP 2E1, providing many additional interesting aspects [110]. For instance, there is a close association between the induction of CYP 2E1 and CYP 2A5/2A6 following prolonged alcohol use that may have an impact on ALD development. Under discussion is also the potential role of CYP 2A5 in ALD including liver fibrosis. Like CYP 2E1, CYP 2A5 is also found in mitochondria but it is still unclear whether alcohol can cause an induction of mitochondrial CYP 2A5 [110]. Interesting but outside of the liver injury issue is the observation that alcohol induction of CYP 2A5 will accelerate blood nicotine clearance and increase the amount of tobacco smoking necessary for maintaining blood nicotine levels. This could explain why patients with an alcohol problem often are confronted with a smoking problem.
Conclusions
Alcohol consumption leads to alcoholic liver disease upon prolonged use in high amounts, causally not associated with the relatively inert ethanol itself but attributable to toxic byproducts generated during enzymatic ethanol oxidation. Apart from alcohol dehydrogenase (ADH), hepatic metabolism proceeds also via the microsomal ethanol oxidizing system (MEOS), which is dependent upon cytochrome P450 (CYP) with a preference of the CYP 2E1 isoform and molecular oxygen. During alcohol metabolism via MEOS and CYP 2E1 and due to incomplete split of oxygen, microsomal oxidative stress develops due to the generation of reactive oxygen species (ROS), which modifies the hepatic immune systems and contributes to the initiation and progression of ALD. A partial role can also be attributed to mitochondrial oxidative stress by CYP 2E1 residing in liver mitochondria and to CYP 2E1 in the mucosa of the intestinal tract, closely associated with the production of endotoxins provided by the gut microbiome. As a result, MEOS, CYP 2E1, and ROS leading to microsomal oxidative stress are largely involved in triggering ALD via radical based toxic effects and alterations of the immune system, but liver mitochondrial and intestinal CYP 2E1 may be contributory in the context of mitochondrial or intestinal oxidative stress.
Despite the abundance of data currently available on MEOS, CYP, and ROS, there are remaining questions. They relate to the mechanisms of CYP dependent ROS generation under hypoxic conditions in face of low oxygen tensions in the centrilobular region of the liver and the consumption of oxygen by CYP 2E1 and MEOS activity. Under clinical aspects, the centrilobular reagion is most vulnerable for injury by alcohol, shown by GDH found in the blood and early appearance of perivenular sclerosis as a precursor of liver fibrosis. There is also uncertainty about how much of the generated ROS is used for microsomal oxidation and remaining for injurious effects. The role of nonCYP2E1 isoforms like CYP 2A5/2A6 in ALD will need further studies. For patients and individuals at risk, validity studies on potential diagnostic biomarkers such as antibodies, blood CYP exosomes, GDH, and endotoxins should be promoted. New therapy studies with inhibitors of CYP 2E1 using phytochemicals or other, nature-based substances can be considered, although preference should be given to alcohol abstinence as the best therapeutical option and to early recognition and prevention of alcohol abuse.
Authors´Contribution
RT provided the first draft after collecting from the coauthor’s valuable suggestions for the outline and receiving papers relevant to the topic. All authors edited the draft, provided substantial intellectual input, and approved the final version to be submitted.
Conflict of Interests
RT declares that he may have a potential conflict of interest regarding this invited article, as he is a member of the editorial board of Archives of Gastroenterology Research. However, he was not involved in the selection of independent reviewers and the final decision regarding acceptance of publication, all done by the editorial office. The other authors declared having no conflict of interests.
References
2. Lieber CS. Effects of ethanol upon lipid metabolism. Lipids. 1974 Feb 1;9(2):103-16.
3. Lieber CS. Alcohol and the liver: 1994 update. Gastroenterology. 1994 Apr 1;106(4):1085-105.
4. Tsukamoto H, Lu SC. Current concepts in the pathogenesis of alcoholic liver injury. The FASEB Journal. 2001 Jun;15(8):1335-49.
5. O’Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Am J Gastroenterol. 2010 Jan;105(1):14-32.
6. Osna NA, Donohue Jr TM, Kharbanda KK. Alcoholic liver disease: Pathogenesis and current management. Alcohol Research: Current Reviews. 2017;38(2):147-161.
7. Seitz HK, Bataller R, Cortez-Pinto H, Gao B, Gual A, Lackner C, et al. Alcoholic liver disease. Nature Reviews Disease Primers. 2018 Aug 16;4(1):16.
8. Neuman MG, Seitz HK, French SW, Malnick S, Tsukamoto H, Cohen LB, et al. Alcoholic-Hepatitis, Links to Brain and Microbiome: Mechanisms, Clinical and Experimental Research. Biomedicines. 2020 Mar;8(3):63.
9. Hasumura Y, Teschke R, Lieber CS. Acetaldehyde oxidation by hepatic mitochondria: decrease after chronic ethanol consumption. Science. 1975 Aug 29;189(4204):727-9.
10. Hasumura Y, Teschke R, Lieber CS. Characteristics of acetaldehyde oxidation in rat liver mitochondria. Journal of Biological Chemistry. 1976 Aug 25;251(16):4908-13.
11. Kubiak-Tomaszewska G, Tomaszewski P, Pachecka J, Struga M, Olejarz W, Mielczarek-Puta M, et al. Molecular mechanisms of ethanol biotransformation: enzymes of oxidative and nonoxidative metabolic pathways in human. Xenobiotica. 2020; 50:1180-1201.
12. Lieber CS, DeCarli LM. Ethanol oxidation by hepatic microsomes: adaptive increase after ethanol feeding. Science. 1968 Nov 22;162(3856):917-8.
13. Lieber CS, DeCarli LM. Hepatic microsomal ethanoloxidizing system in vitro characteristics and adaptive properties in vivo. Journal of Biological Chemistry. 1970 May 25;245(10):2505-12.
14. Lieber CS, DeCarli LM, Matsuzaki S, Ohnishi K, Teschke R. The microsomal ethanol oxidizing system. In: Methods in Enzymology; Fleischer, S., Packer, L., Eds.; Academic Press: New York, NY, USA, 1978; pp. 355-368.
15. Teschke R, Hasumura Y, Joly JG, Ishii H, Lieber CS. Microsomal ethanol-oxidizing system (MEOS): Purification and properties of a rat liver system free of catalase and alcohol dehydrogenase. Biochemical and Biophysical Research Communications. 1972 Dec 4;49(5):1187-93.
16. Teschke R, Hasumura Y, Lieber CS. NADPHdependent oxidation of methanol, ethanol, propanol and butanol by hepatic microsomes. Biochemical and Biophysical Research Communications. 1974 Sep 23;60(2):851-7.
17. Teschke R, Hasumura Y, Lieber CS. Hepatic microsomal ethanol-oxidizing system: solubilization, isolation, and characterization. Archives of Biochemistry and Biophysics. 1974 Jul 1;163(1):404-15.
18. Teschke R, Hasumura Y, Lieber CS. Hepatic microsomal alcohol-oxidizing system. Affinity for methanol, ethanol, propanol, and butanol. Journal of Biological Chemistry. 1975 Sep 25;250(18):7397-404.
19. Teschke R, Ohnishi K, Hasumura Y, Lieber CS. Hepatic microsomal ethanol oxidizing system: Isolation and reconstitution. In: Microsomes and Drug Oxidations 1977 Jan 1 (pp. 103-110). Pergamon.
20. Ohnishi KU, Lieber CS. Reconstitution of the microsomal ethanol-oxidizing system. Qualitative and quantitative changes of cytochrome P-450 after chronic ethanol consumption. Journal of Biological Chemistry. 1977 Oct 25;252(20):7124-31.
21. Lieber CS, DeCarli LM. The role of the hepatic microsomal ethanol oxidizing system (MEOS) for ethanol metabolism in vivo. Journal of Pharmacology and Experimental Therapeutics. 1972 May 1;181(2):279-87.
22. Teschke R. Alcoholic liver disease: alcohol metabolism, cascade of molecular mechanisms, cellular targets, and clinical aspects. Biomedicines. 2018 Dec;6(4):106.
23. Teschke R. Alcoholic liver disease: Current mechanistic aspects with focus on their clinical relevance. Biomedicines. 2019 Sep;7(3):68.
24. Teschke R. Microsomal ethanol-oxidizing system: success over 50 years and an encouraging future. Alcoholism: Clinical and Experimental Research. 2019 Mar;43(3):386-400.
25. García-Bañuelos J, Panduro A, Gordillo-Bastidas D, Gordillo-Bastidas E, Muñoz-Valle JF, Gurrola-Díaz CM, et al. Genetic polymorphisms of genes coding to alcoholmetabolizing enzymes in Western Mexicans: association of CYP2E1* c2/CYP2E1* 5B allele with cirrhosis and liver function. Alcoholism: Clinical and Experimental Research. 2012 Mar;36(3):425-31.
26. Gaviria-Calle M, Duque-Jaramillo A, Aranzazu M, Filippo DD, Montoya M, Roldán I, Palacio N, et al. Polymorphisms in alcohol dehydrogenase (ADH1) and cytochrome p450 2E1 (CYP2E1) genes in patients with cirrhosis and / or hepatocellular carcinoma. Biomedical. 2018 Dec; 38 (4): 555-68.
27. Jiang Y, Zhang T, Kusumanchi P, Han S, Yang Z, Liangpunsakul S. Alcohol metabolizing enzymes, microsomal ethanol oxidizing system, cytochrome P450 2E1, catalase, and aldehyde dehydrogenase in alcoholassociated liver disease. Biomedicines. 2020 Mar;8(3):50.
28. Blacker TS, Duchen MR. Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radical Biology and Medicine. 2016 Nov 1;100:53-65.
29. Teschke R, Hasumura Y, Lieber CS. Hepatic microsomal alcohol-oxidizing system in normal and acatalasemic mice: its dissociation from the peroxidatic activity of catalase-H2O2. Molecular Pharmacology. 1975 Nov 1;11(6):841-9.
30. Teschke R. Biochemical aspects of the hepatic microsomal ethanol-oxidizing system (MEOS): Resolved initial controversies and updated molecular views. Biochem. Pharmacol. 2019;8(267):2167-0501.
31. Rubin E, Lieber CS. Early fine structural changes in the human liver induced by alcohol. Gastroenterology. 1967 Jan 1;52(1):1-13.
32. Rubin E, Hutterer F, Lieber CS. Ethanol increases hepatic smooth endoplasmic reticulum and drug-metabolizing enzymes. Science. 1968 Mar 29;159(3822):1469-70.
33. Joly JG, Ishii H, Teschke R, Hasumura Y, Lieber CS. Effect of chronic ethanol feeding on the activities and submicrosomal distribution of reduced nicotinamide adenine dinucleotide phosphate-cytochrome P-450 reductase and the demethylases for aminopyrine and ethylmorphine. Biochemical Pharmacology. 1973 Jun 15;22(12):1532-5.
34. Miwa GT, Levin W, Thomas PE, Lu AY. The direct oxidation of ethanol by a catalase-and alcohol dehydrogenase-free reconstituted system containing cytochrome P-450. Archives of Biochemistry and Biophysics. 1978 Apr 30;187(2):464-75.
35. Damgaard SE. The D (V/K) isotope effect of the cytochrome P-450-mediated oxidation of ethanol and its biological applications. European Journal of Biochemistry. 1982 Jul;125(3):593-603.
36. Lu Y, Cederbaum AI. CYP2E1 and oxidative liver injury by alcohol. Free Radical Biology and Medicine. 2008 Mar 1;44(5):723-38.
37. Neve EP, Ingelman-Sundberg M. Identification and characterization of a mitochondrial targeting signal in rat cytochrome P450 2E1 (CYP2E1). Journal of Biological Chemistry. 2001 Apr 6;276(14):11317-22.
38. Ingelman-Sundberg M. A soluble NH(2)-terminally truncated catalytically active form of rat cytochrome P450 2E1 targeted to liver mitochondria(1). FEBS Lett. 1999;460:309-314.
39. Robin MA, Anandatheerthavarada HK, Fang JK, Cudic M, Otvos L, Avadhani NG. Mitochondrial targeted cytochrome P450 2E1 (P450 MT5) contains an intact N terminus and requires mitochondrial specific electron transfer proteins for activity. Journal of Biological Chemistry. 2001 Jul 6;276(27):24680-9.
40. Robin MA, Anandatheerthavarada HK, Biswas G, Sepuri NB, Gordon DM, Pain D, et al. Bimodal targeting of microsomal CYP2E1 to mitochondria through activation of an N-terminal chimeric signal by cAMP-mediated phosphorylation. Journal of Biological Chemistry. 2002 Oct 25;277(43):40583-93.
41. Robin MA, Sauvage I, Grandperret T, Descatoire V, Pessayre D, Fromenty B. Ethanol increases mitochondrial cytochrome P450 2E1 in mouse liver and rat hepatocytes. FEBS letters. 2005 Dec 19;579(30):6895-902.
42. Asai H, Imaoka S, Kuroki T, MONNA T, Funae Y. Microsomal ethanol oxidizing system activity by human hepatic cytochrome P450s. The Journal of Pharmacology and Experimental Therapeutics. 1996;277(2): 1004-1009.
43. Lieber CS. Microsomal ethanol-oxidizing system (MEOS): the first 30 years (1968-1998)–a review. Alcoholism: Clinical and Experimental Research. 1999 Jun;23(6):991-1007.
44. Lieber CS. Cytochrome P-4502E1: its physiological and pathological role. Physiological Reviews. 1997 Apr 1;77(2):517-44.
45. Lieber CS. The discovery of the microsomal ethanol oxidizing system and its physiologic and pathologic role. Drug Metabolism Reviews. 2004 Jan 1;36(3-4):511-29.
46. Kessova I, Cederbaum AI. CYP2E1: biochemistry, toxicology, regulation and function in ethanol-induced liver injury. Current Molecular Medicine. 2003 Sep 1;3(6):509-18.
47. Ingelman-Sundberg M, Johansson I. Mechanisms of hydroxyl radical formation and ethanol oxidation by ethanol-inducible and other forms of rabbit liver microsomal cytochromes P-450. Journal of Biological Chemistry. 1984 May 25;259(10):6447-58.
48. Ingelman-Sundberg M, Johansson I, Yin H, Terelius Y, Eliasson E, Clot P, et al. Ethanol-inducible cytochrome P4502E1: genetic polymorphism, regulation, and possible role in the etiology of alcohol-induced liver disease. Alcohol. 1993 Nov 1;10(6):447-52.
49. Albano E, Tomasi A, Ingelman-Sundberg M. [11] Spin trapping of alcohol-derived radicals in microsomes and reconstituted systems by electron spin resonance. In: Methods in Enzymology 1994 Jan 1 (Vol. 233, pp. 117-127).
50. Ingelman-Sundberg M, Johansson I. Mechanisms of hydroxyl radical formation and ethanol oxidation by ethanol-inducible and other forms of rabbit liver microsomal cytochromes P-450. Journal of Biological Chemistry. 1984 May 25;259(10):6447-58.
51. Ingelman-Sundberg M, Jörnvall H. Induction of the ethanol-inducible form of rabbit liver microsomal cytochrome P-450 by inhibitors of alcohol dehydrogenase. Biochemical and Biophysical Research Communications. 1984 Oct 30;124(2):375-82.
52. Ingelman-Sundberg M, Hagbjörk AL. On the significance of the cytochrome P-450-dependent hydroxyl radical-mediated oxygenation mechanism. Xenobiotica. 1982 Jan 1;12(11):673-86.
53. Cederbaum AI. Oxygen radical generation by microsomes: role of iron and implications for alcohol metabolism and toxicity. Free Radical Biology and Medicine. 1989 Jan 1;7(5):559-67.
54. Cederbaum AI. Molecular mechanisms of the microsomal mixed function oxidases and biological and pathological implications. Redox Biology. 2015 Apr 1;4:60-73.
55. Korsten MA, Matsuzaki S, Feinman L, Lieber CS. High blood acetaldehyde levels after ethanol administration: Difference between alcoholic and nonalcoholic subjects. New England Journal of Medicine. 1975 Feb 20;292(8):386-9.
56. Davis VE, Walsh MJ. Alcohol, amines, and alkaloids: a possible biochemical basis for alcohol addiction. Science. 1970 Feb 13:1005-7.
57. Cohen G, Cederbaum AI. Microsomal metabolism of hydroxyl radical scavenging agents: relationship to the microsomal oxidation of alcohols. Archives of Biochemistry and Biophysics. 1980 Feb 1;199(2):438-47.
58. Ohnishi K, Lieber CS. Respective role of superoxide and hydroxyl radical in the activity of the reconstituted microsomal ethanol-oxidizing system. Archives of Biochemistry and Biophysics. 1978 Dec 1;191(2):798-803.
59. Winston GW, Cederbaum AI. Evidence for two ethanol oxidizing pathways in reconstituted mixedfunction oxidase systems. Pharmacology Biochemistry and Behavior. 1983 Jan 1;18:189-94.
60. Mello T, Zanieri F, Ceni E, Galli A. Oxidative stress in the healthy and wounded hepatocyte: a cellular organelles perspective. Oxidative Medicine and Cellular Longevity. 2016 Jan 1;2016.
61. Milic S, Mikolasevic I, Orlic L, Devcic E, Starcevic- Cizmarevic N, Stimac D, et al. The role of iron and iron overload in chronic liver disease. Medical science monitor: International Medical Journal of Experimental and Clinical Research. 2016;22:2144-51.
62. Duryee MJ, Klassen LW, Thiele GM. Immunological response in alcoholic liver disease. World Journal of Gastroenterology. 2007 Oct 7;13(37):4938-46.
63. Albano E, Vidali M. Immune mechanisms in alcoholic liver disease. Genes & Nutrition. 2010 Jun 1;5(2):141-7.
64. Nagy LE. The role of innate immunity in alcoholic liver disease. Alcohol Research: Current Reviews. 2015; 37: 237-250.
65. Pasala S, Barr T, Messaoudi I. Impact of alcohol abuse on the adaptive immune system. Alcohol Research: Current Reviews. 2015;37(2):185-197.
66. Li S, Tan HY, Wang N, Feng Y, Wang X, Feng Y. Recent Insights into the Role of Immune Cells in Mediating Inflammation of Alcoholic Liver Disease. Frontiers in Immunology. 2019;10:1328.
67. Uetrecht J. Mechanistic studies of idiosyncratic DILI: Clinical implications. In: Special issue: Clinical drug induced liver injury: Current diagnostic and mechanistic challenges. Guest editors: Rolf Teschke, Gaby Danan & James H. Lewis. Frontiers in Pharmacology. 2019; 10: 3389
68. Teschke R, Uetrecht J. Mechanism of idiosyncratic drug induced liver injury (DILI): Unresolved basic issues. In special issue: Unresolved basic issues in hepatology, guest editors Ralf Weiskirchen & Wolfgang Stremmel. Annals of Translational Medicine 2020.
69. Uetrecht J, Naisbitt DJ. Idiosyncratic adverse drug reactions: current concepts. Pharmacological Reviews. 2013 Apr 1;65(2):779-808.
70. Uetrecht J. Are drugs containing carboxylic acid functional group associated with a significant risk of idiosyncratic drug reactions? Journal of Modern Medicinal Chemistry. 2020; 8: 56-64
71. Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nature Immunology. 2015 Apr;16(4):343-53.
72. Medzhitov R, Janeway Jr C. Innate immune recognition: mechanisms and pathways. Immunological Reviews. 2000 Feb;173:89-97.
73. Teschke R. Alcoholic steatohepatitis (ASH) and alcoholic hepatitis (AH): cascade of events, clinical aspects, and pharmacotherapy options. Expert Opinion on Pharmacotherapy. 2018 May 24;19(8):779-93.
74. Trump BF, Duttera SM, Byrne WL, Arstila AU. Membrane structure: lipid-protein interactions in microsomal membranes. Proceedings of the National Academy of Sciences. 1970 Jun 1;66(2):433-40.
75. Colbeau A, Nachbaur J, Vignais PM. Enzymac characterization and lipid composition of rat liver subcellular membranes. Biochimica et Biophysica Acta (BBA)-Biomembranes. 1971 Dec 3;249(2):462-92.
76. Laganiere S, Byung PY. Modulation of membrane phospholipid fatty acid composition by age and food restriction. Gerontology. 1993;39(1):7-18.
77. Bode JC, Bode C, Heidelbach R, Dürr HK, Martini GA. Jejunal microflora in patients with chronic alcohol abuse. Hepato-Gastroenterology. 1984 Feb;31(1):30-34.
78. Bode C, Kugler V, Bode JC. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. Journal of Hepatology. 1987 Jan 1;4(1):8- 14.
79. Fukui H, Brauner B, Bode JC, Bode C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic assay. Journal of Hepatology. 1991 Mar 1;12(2):162-9.
80. Bode C, Kolepke R, Schäfer K, Bode JC. Breath hydrogen excretion in patients with alcoholic liver disease--evidence of small intestinal bacterial overgrowth. Zeitschrift für Gastroenterologie. 1993 Jan;31(1):3-7.
81. Schäfer C, Parlesak A, Schütt C, Christian Bode J, Bode C. Concentrations of lipopolysaccharide-binding protein, bactericidal/permeability-increasing protein, soluble CD14 and plasma lipids in relation to endotoxaemia in patients with alcoholic liver disease. Alcohol and Alcoholism. 2002 Jan 1;37(1):81-6.
82. Bode C, Bode JC. Effect of alcohol consumption on the gut. Best Practice & Research Clinical Gastroenterology. 2003 Aug 1;17(4):575-92.
83. Purohit V, Bode JC, Bode C, Brenner DA, Choudhry MA, Hamilton F, et al. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: summary of a symposium. Alcohol. 2008 Aug 1;42(5):349-61.
84. Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011 Nov 1;141(5):1572-85.
85. Dunn W, Shah VH. Pathogenesis of alcoholic liver disease. Clinics in Liver Disease. 2016 Aug 1;20(3):445-56.
86. Szabo G. Gut–liver axis in alcoholic liver disease. Gastroenterology. 2015 Jan 1;148(1):30-6.
87. Szabo G, Petrasek J. Gut–liver axis and sterile signals in the development of alcoholic liver disease. Alcohol and Alcoholism. 2017 Jul 1;52(4):414-24.
88. Neuman MG, French SW, Zakhari S, Malnick S, Seitz HK, Cohen LB, et al. Alcohol, microbiome, life style influence alcohol and non-alcoholic organ damage. Experimental and Molecular Pathology. 2017 Feb 1;102(1):162-80.
89. Neuman MG, French SW, French BA, Seitz HK, Cohen LB, Mueller S, et al. Alcoholic and non-alcoholic steatohepatitis. Experimental and Molecular Pathology. 2014 Dec 1;97(3):492-510.
90. Méndez-Sánchez N, Valencia-Rodriguez A, Vera- Barajas A, Abenavoli L, Scarpellini E, Ponciano-Rodriguez G, et al. The mechanism of dysbiosis in alcoholic liver disease leading to liver cancer. Hepatoma Research. 2020;6: 5.
91. Seitz HK, Korsten MA, Lieber CS. Ethanol oxidation by intestinal microsomes: increased activity after chronic ethanol administration. Life Sciences. 1979 Oct 15;25(16):1443-8.
92. Seitz HK, Bösche J, Czygan P, Veith S, Kommerell B. Microsomal ethanol oxidation in the colonic mucosa of the rat. Naunyn-Schmiedeberg’s Archives of Pharmacology. 1982 Mar 1;320(1):81-4.
93. Bergheim I, Bode C, Parlesak A. Distribution of cytochrome P450 2C, 2E1, 3A4, and 3A5 in human colon mucosa. BMC Clinical Pharmacology. 2005 Dec 1;5(1):4.
94. Abdelmegeed MA, Banerjee A, Jang S, Yoo SH, Yun JW, Gonzalez FJ, et al. CYP2E1 potentiates binge alcoholinduced gut leakiness, steatohepatitis, and apoptosis. Free Radical Biology and Medicine. 2013 Dec 1;65:1238-45.
95. Forsyth CB, Voigt RM, Keshavarzian A. Intestinal CYP2E1: A mediator of alcohol-induced gut leakiness. Redox Biology. 2014 Jan 1;3:40-6.
96. Teschke R, Zhu Y. Opinion: Intestinal microbiome, endotoxins, cytochrome P450 2E1, and the gut-liver axis in alcoholic liver disease. EC Gastroenterology Digestive Systems. 2019; 5, 11.
97. Summa KC, Voigt RM, Forsyth CB, Shaikh M, Cavanaugh K, Tang Y, Vitaterna MH, Song S, Turek FW, Keshavarzian A. Disruption of the circadian clock in mice increases intestinal permeability and promotes alcoholinduced hepatic pathology and inflammation. PloS One. 2013 Jun 18;8(6):e67102.
98. Engen PA, Green SJ, Voigt RM, Forsyth CB, Keshavarzian A. The gastrointestinal microbiome: alcohol effects on the composition of intestinal microbiota. Alcohol Research: Current Reviews. 2015;37(2):223-36.
99. Pirmohamed M, Kitteringham NR, Quest LJ, Allott RL, Green VJ, Gilmore IT, et al. Genetic polymorphism of cytochrome P4502E1 and risk of alcoholic liver disease in Caucasians. Pharmacogenetics. 1995 Dec 1;5(6):351-7.
100. Morgan K, French SW, Morgan TR. Production of a cytochrome P450 2E1 transgenic mouse and initial evaluation of alcoholic liver damage. Hepatology. 2002 Jul;36(1):122-34.
101. Lu Y, Wu D, Wang X, Ward SC, Cederbaum AI. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knockin mice. Free Radical Biology and Medicine. 2010 Nov 15;49(9):1406-16.
102. Gebhardt AC, Lucas D, Menez J, Seitz HK. Chlormethiazole inhibition of cytochrome P450 2E1 as assessed by chlorzoxazone hydroxylation in humans. Hepatology. 1997 Oct;26(4):957-61.
103. Gouillon ZQ, Lucas D, Li J, Hagbjork AL, French BA, Fu P, et al. Inhibition of Ethanol-Induced Liver Disease in the Intragastric Feeding Rat Model by Chlormethiazole. Proceedings of the Society for Experimental Biology and Medicine. 2000 Apr;224(4):302-8.
104. Teschke R, Xuan TD. Active nature-based ingredients for drug discovery with pivotal role of clinical efficacy: review and prospective. Journal of Modern Medicinal Chemistry. 2020;8:4-18.
105. Cho YE, Mezey E, Hardwick JP, Salem Jr N, Clemens DL, Song BJ. Increased ethanol-inducible cytochrome P450-2E1 and cytochrome P450 isoforms in exosomes of alcohol-exposed rodents and patients with alcoholism through oxidative and endoplasmic reticulum stress. Hepatology Communications. 2017 Sep;1(7):675-90.
106. Teschke R, Eickhoff A, Brown AC, Neuman MG, Schulze J. Diagnostic biomarkers in liver injury by drugs, herbs, and alcohol: tricky dilemma after EMA correctly and officially retracted letter of support. International Journal of Molecular Sciences. 2020 Jan;21(1):212.
107. Van Waes L, Lieber CS. Glutamate dehydrogenase: a reliable marker of liver cell necrosis in the alcoholic. British Medical Journal. 1977 Dec 10;2(6101):1508-10.
108. Worner TM, Lieber CS. Plasma glutamate dehydrogenase: Clinical application in patients with alcoholic liver disease. Alcoholism: Clinical and Experimental Research. 1980 Oct;4(4):431-4.
109. Ingelman-Sundberg M, Johansson I, Penttilä KE, Glaumann H, Lindros KO. Centrilobular expression of ethanol-inducible cytochrome P-450 (IIE1) in rat liver. Biochemical and Biophysical Research Communications. 1988 Nov 30;157(1):55-60.
110. Lu Y, Cederbaum AI. Cytochrome P450s and alcoholic liver disease. Current Pharmaceutical Design. 2018 Apr 1;24(14):1502-17.