Letter to the Editor
In mature B-cells, class switch recombination (CSR) substitutes the constant (C) μ gene with other C genes (such as γ, ε and α) thereby generating IgG, IgE and IgA antibodies with new effector functions but same the antigenic specificity compared to IgM. CSR is a complex process with I-promoter transcription, targeting of the DNA-editing enzyme activation-induced deaminase (AID) to specific DNA switch (S) regions preceding C regions (except Cδ), generation of double strand breaks and recruitment of DNA repair factors for the end-joining process. The classical non-homogenous end joining (c-NHEJ) pathway ligates DNA ends with little or no homology while the alternative end joining (A-EJ) pathway ligates DNA ends with microhomology [1]. Several cis-transcriptional enhancers located along the IgH locus have been implicated (near or far) in CSR control. Deletion of the 3’ regulatory region (3’RR) (located 3’ to the Cα gene) markedly reduced conventional (γ, ε and α) CSR [2]. Deletion of the 5’Eμ enhancer (located upstream of Cμ) has, at most, a modest effect on synthesis of switched Ig and no really evident role on CSR [3]. Deletion of the 3’γ1E element (located between Cγ1 and Cγ2b genes) restricts CSR to IgG3, IgG2a and IgG2b illustrating its role as an isotype specific transcriptional enhancer of CSR [4]. IgD CSR is a rare and enigmatic CSR restricted to a few B-cell subsets in specific lymphoid tissues (such as mucosaassociated tissue, mesenteric lymph nodes and the peritoneal cavity) [5,6]. A link between IgD CSR and microbiota has been reported demonstrating a role for IgD in the homeostatic regulation of the microbial community [7]. The mechanistic regulation of IgD CSR remains elusive. Transcription is a prerequisite for conventional CSR and deletion of several IgH cis-enhancers are reported to affect it. AID targeting on S regions is a prerequisite for conventional CSR and AID deletion abolishes it. We investigated the role of IgH cis-enhancers and members of the AID/APOBEC family during IgD CSR.
Eμ-deficient [3], 3’Eγ1-deficient [4], APOBEC3-deficient [8], AID-deficient (Janvier Labs, France), wt 129/SV and wt C57BL6 mice were housed and procedures were conducted in agreement (APAFIS-13855) with European Directive 2010/63/ EU on animals used for scientific purposes. Peritoneal cavity B-cells were used. Mice were pristane-treated (1ml i.p.) for 2 weeks to induce inflammation before recovery of peritoneal cavity cells. B-cells were purified using EasySep™ mouse B-cell isolation Kit (STEMCELL Technologies). DNA was extracted by phenol/chloroform. Sμ-σδ (σ for S-like) junctions were amplified by nested PCR with 100 ng DNA (Phusion High-Fidelity PCR Master Mix with HF Buffer) as previously reported [9]. NGS Barcoded libraries with 200-bp read lengths were prepared using Ion Xpress plus Fragment Library Kit (Thermo Fisher Scientific) according to manufacturer’s instructions. 100 pM of each barcoded library were run on the Ion Proton sequencer (Life Technologies) located in the “GénoLim platform” of the Limoges University (France). Data analysis was performed using the computational tool for automatic analysis of CSR junctions sequenced by high-throughput sequencing (CSReport) [10]. Sequenced reads were mapped to Sμ and σδ regions using a BLAST algorithm. The computational tool developed for experiments performs junction assembly, identifies breakpoints in Sμ and σδ, identifies junction structure (blunt, micro-homology, large-homology or junction with insertions) and outputs a statistical summarization of identified junctions.
The IgH locations of Sμ, σδ, Cμ, Cδ, 5’Eμ, 3’Eγ1 and 3’RR are depicted in Figure 1A. If transcription is required for CSR, we previously reported that deletion of the 3’RR master control element of CSR had no effect on IgD CSR [6]. Since 3’RR is far from σδ-Cδ we investigated the impact of deletion of the two other nearby cis-transcriptional elements 5’Eμ (mice with a 129 background) and 3’Eγ1 (mice with a C57BL6 background). IgD CSR was previously reported in 129/SV and C57BL6 wt mice [6,7]. Data indicated the presence of IgD CSR in 5’Eμ- and 3’Eγ1- deficient mice (Figure 1B). Analysis of Sμ-σδ junctions indicated that their structural profile (blunt, micro-homology, largehomology or junction with insertions) (Figure 1B) was not significantly different from IgD junctions in 129/SV and C57BL6 wt mice. Thus, deletion of the 3 major cis-transcriptional enhancers of the IgH locus did not affect IgD CSR suggesting that cis-enhancer-induced transcription is not required for this non-conventional CSR. Comparison of IgD junctions with IgG3 junctions (obtained after LPS stimulation) or IgG1 junctions (obtained after LPS+IL4 stimulation) revealed no significant differences in their structural profile (Figure 1C) suggesting a similar end-joining process for δ, γ3 and γ1 CSR and that mechanistic differences between IgD CSR and conventional CSR occurred in the first step of the CSR process. AID is essential for conventional CSR and colocation of breakpoints in Sμ with AID hot spot motifs was found in IgD junctions [9]. Confirming the involvement of AID in IgD CSR, no significant numbers of Sμ-σδ junctions were found in AID-deficient mice (Figure 1D). Analysis of breakpoints in σδ collocated with motifs mostly targeted by APOBEC3 [9]. Confirming the putative involvement of APOBEC3 in IgD CSR, a significant decrease in the number of Sμ-σδ junctions was found in APOBEC3-deficient mice (Figure 1D). Of interest, IgD junction levels increased in APOBEC3-deficient mice expressing a human APOBEC3A or APOBEC3G transgene (a single APOBEC3 gene is present in rodents whereas seven copies are found in humans). The Sμ- σδ junctions in human APOBEC3A/G expressing mice were slightly different from those of wt control mice with less blunt junctions and more junctions with short microhomologies (Figure 1E).
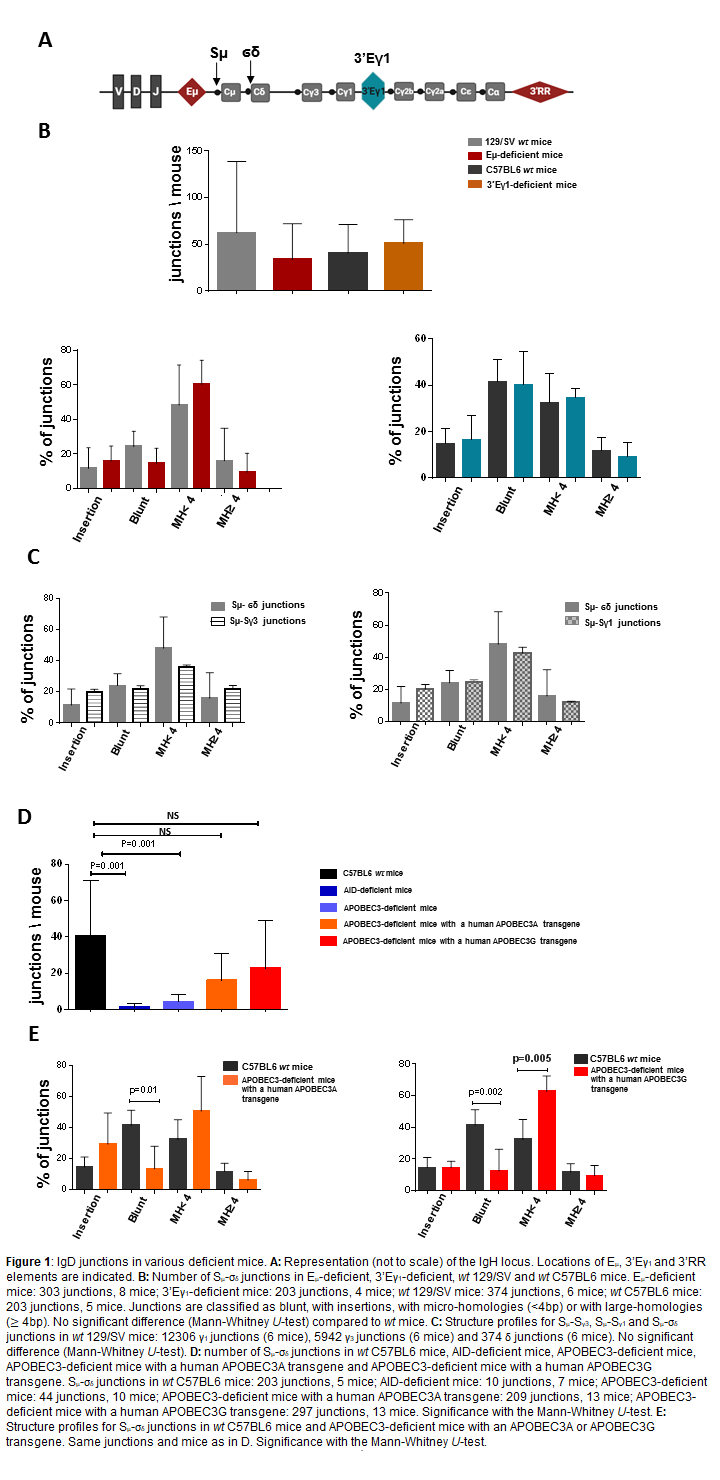
In mature B-cells the expression of IgM and IgD through alternative splicing of a long mRNA transcript is constitutive indicating that Sμ and σδ regions are always transcribed and explaining why no cis-enhancer-induced transcription (3’RR, 5’Eμ, 3’Eγ1) is required for IgD CSR. Similarly, for conventional CSR, AID is clearly implicated in IgD CSR, most probably through its constant Sμ targeting. APOBEC3 proteins act in cancer and in gene editing (including Ig ones) [11,12]. When IgD CSR is almost totally abrogated in AID-deficient mice (AID is essential to target Sμ), this was not the case in an APOBEC3-deficient background where a residual level of Sμ-σδ junctions was found. An explanation would be that APOBEC3 hot spots in σδ are also AID cold spots. Expression of human APOBEC3A/G transgenes (both expressed in human B-cells) in an APOBEC3-deficient background increased levels of Sμ- σδ junctions but with a different panel of junctions that could be explained by the fact that mouse APOBEC3 preferentially deaminates TYC motifs (Y=T/C) whereas TC and CCC motifs are preferred by human APOBEC3A and APOBEC3G, respectively [13]. Recent discoveries of IgD in ancient vertebrates and AID/APOBEC-like cytidine deaminases as ancient innate immune mediators in invertebrates suggest that IgD has been preserved throughout evolution [14]. IgD CSR would be an evolving residue allowing the specific production of IgD, which is reactive to commensal microbiota, before expansion of the IgH locus by duplication [15], leading to the emergence of γ, α and ε isotypes and subsequently conventional AIDinduced CSR induced via the appearance of conventional GCrich S regions. Clearly, the role for APOBEC3 in the targeting of the σδ region during the IgD CSR deserves more studies. The difficulty to monitor it, due to the rarity of this event which is restricted to a few B-cell subsets in specific mouse lymphoid tissues and currently impossible to reproduce in vitro, remains an exciting challenge to be met.
Acknowledgments
Authors are “Equipe Labellisée LIGUE 2018”. This work was supported by the ANR (project EpiSwitch-3’RR 2016). N.G. and H.I. were supported by a grant from ANR. M.F. was supported by University of Limoges and “Région Nouvelle Aquitaine”. We thank Bernardo Reina-San-Martin (IGBMC, Illkirch, France) for sending us 3’Eγ1-deficient mice.
Disclosure of Conflicts of Interest
The authors declare no conflict of interest.
Author Contributions
Y.D and J.C.M. designed research. M.F., N.G. and H.I. performed research. M.F., J.C.M. and Y.D. analyzed data. M.F., N.G., H.I., J.C.M., and Y.D. wrote the paper. Y.D. obtained financial grants.
References
2. Vincent-Fabert C, Fiancette R, Pinaud E, Truffinet V, Cogné, Cogné M, et al. Genomic deletion of the whole IgH 3’ regulatory region (hs3a, hs1,2, hs3b, hs4) dramatically affects class switch recombination and Ig secretion to all isotypes. Blood. 2010;116:1895-1898.
3. Marquet M, Garot A, Bender S, Carrion C, Rouaud P, Lecardeur S, et al. The Eμ enhancer region influences H chain expression and B cell fate without impacting IgVH repertoire and immune response in vivo. J. Immunol. 2014;193:1171-1183.
4. Amoretti-Villa R, Rogier M, Robert I, Heyer V, Reina-San-Martin B. A novel regulatory region controls IgH locus transcription and switch recombination to a subset of isotypes. Cell Mol Immunol. 2019;16:887-889.
5. Chen K, Xu W, Wilson M, He B, Miller NW, Bengtén E, et al. Immunoglobulin D enhances immune surveillance by activating antimicrobial, proinflammatory and B cell-stimulating programs in basophils. Nat Immunol. 2009;10:889–898.
6. Rouaud P, Saintamand A, Saad F, Carrion C, Lecardeur S, Cogné M, et al. Elucidation of the enigmatic IgD class-switch recombination via germline deletion of the IgH 3’ regulatory region. J Exp Med. 2014;211:975-985.
7. Choi JH, Wang KW, Zhang D, Zhan X, Wang T, Bu CH, et al. IgD class switching is initiated by microbiota and limited to mucosaassociated lymphoid tissue in mice. Proc Natl Acad Sci USA. 2017;114:E1196-E1204.
8. Okeoma CM, Lovsin N, Peterlin BM, Ross SR. APOBEC3 inhibits mouse mammary tumour virus replication in vivo. Nature. 2007;445: 927-930.
9. Ghazzaui N, Issaoui H, Saintamand A, Boyer F, Denizot Y.Analysis of IgD CSR junctions by high-throughput sequencing. Immunol Lett. 2017;188:86-88.
10. Boyer F, Boutouil H, Dalloul I, Dalloul Z, Cook-Moreau J, Aldigier JC, et al. CSReport: a new computational tool designed for automatic analysis of class switch recombination junctions sequenced by high-throughput sequencing. J Immunol. 2017;198:4148-4155.
11. Silvas TV, Schiffer CA. APOBEC3s: DNA-editing human cytidine deaminases. Protein Science. 2019;28:1552-1566.
12. Buisson R, Langenbucher A, Bowen D, Kwan EE, Benes CH, Zou L, et al. Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science. 2019;364:eaaw2872.
13. Shapiro M, Meier S, MacCarthy T. The cytidine deaminase under-representation reporter (CDUR) as a tool to study evolution os sequences under deaminase mutational pressure. BMC Bioinformatics. 2018;19:163.
14. Liu MC, Liao WY, Buckley KM, Yang SY, Rast JP, Fugmann SD. AID/APOBEC-like cytidine deaminases are ancient innate immune mediators in invertebrates. Nat Commun. 2018;9:1948.
15. Magor BG, Ross DA, Pilström L, Warr GW. Transcriptional enhancers and the evolution of the IgH locus. Immunology Today. 1999;20:13-17.