Abstract
Cell signaling pathways involve several proteins, called kinases, to pass on vital messages from the transmembrane-proteins, e.g., receptor tyrosine kinases (RTKs) to the nucleus of a cell through a cascade of processes for various activities such as controlled cell division, proliferation, apoptosis, metabolism, DNA replication and repair. Alterations in these signaling proteins combined with genetic and/or epigenetic mutations spawn various types of cancer that empowers the cells to ignore or maneuver the immune signals and carry on felonious activities such as uncontrolled cell division and growth, genetic instability, angiogenesis around the tumor in hypoxia, eluding the body’s immune system, cell migration, and invasion of surrounding healthy cells. Inhibition of such malicious transmuted signaling pathways has therapeutic importance for cancer. In this article, we have briefly described three vital signaling pathways that are aberrantly triggered by Ras and Wnt proteins and the NF-2 genes causing different types of cancer. Recent research in the targeted therapy using drugs to put off these signaling pathways, especially, for cancer stem cells (CSCs) has been reviewed here. However, such therapy suffers a low success rate due to the resistance to drugs by cancer cells. Considering this snag of targeting drugs, many researchers use nanoparticles for this purpose; some of these works have been reviewed in brief. The consequences of protein-nanoparticles interaction in this approach has also been discussed. Finally, we have proposed use of nanomaterials for targeted ion delivery, or targeted heating using light or magnetic field, to destroy cancer cells.
Keywords
Cancer, Signaling pathways, Targeted therapy, Cancer stem cells, Nanomaterials, Protein-nanoparticle interaction, Cell death
Introduction
Cancer develops due to genetic and/or epigenetic mutations. Some mutations produce oncogenes that drive tumor formation, while others inactivate tumor suppressor genes that control inappropriate cell proliferation and persistence outside their regular niche. These mutations empower cells to bypass homeostatic suppression of inappropriate proliferation, cell migration, and aggressive invasion [1].
Several signaling pathways trigger normal cellular activities. Few membrane proteins such as receptor tyrosine kinases (RTKs) bind extracellular messenger molecules such as hormones or growth factors (called ligands) and undergo a conformational change relaying the signal across the membrane to the receptor’s cytoplasmic domain. Subsequently, the membrane-bound protein is activated (by phosphorylation), for example, Ras in its active GTP-bound state. GTPase-activating proteins such as neurofibromin-1 (NF-1) deactivate (dephosphorylation) Ras by converting it to an inactive GDP-bound state. The activated protein interacts with several downstream effectors initiating cascades of enzymic activities through one of the pathways to regulate cellular activities, like cell division and growth, repair of damaged DNA, glycolysis, and apoptosis [2,3]. Dysregulation in these signal transduction pathways boons the cell’s capacity to proliferate independently of exogenous growthpromoting or growth-inhibitory signals, invade surrounding tissues and metastasize to distant sites, to resist apoptosis and other forms of cell death, metabolic activities in hypoxia, sustain with genetic instability, and to induce angiogenesis [4]. Proteins associated with these dysregulated pathways are currently under investigation as possible targets of various drugs to develop cancer therapy. In this article, we have reviewed three cell signaling pathways triggered by Ras and Wnt proteins and the NF-2 genes in connection with different types of cancer and accounted for recent progress in targeted therapy of these signaling pathways using drugs. Finally, we have administered for the nanomaterials to target aberrant signaling pathways in cancer cells.
Oncogenic Mutations and Aberrant Signaling Pathways
Here we highlight the mutation of two proteins and a gene that can exemplify aberrantly activated signaling pathways for cancer development. Mutations that convert cellular proto-oncogenes to oncogenes can cause hyperactivation of the signaling pathways, whereas inactivation of tumor suppressors eliminates critical negative regulators of signaling [5]. The following examples illustrate dysregulated signaling pathways in cancer.
Ras protein
The Ras is a G-protein that acts as a molecular switch to control the activation and regulation of two pathways of Raf-MEK-ERK and PI3K-AKT cell signaling through which it regulates cell survival and proliferation, migration, and invasion [6,7]. Figure 1 shows a simple schematic diagram of these two pathways. Raf, mitogen-activated protein kinase (MAPK), extracellular receptor kinase (ERK), AKT, and mammalian target of rapamycin (mTOR) come under serine/ threonine-specific protein kinases (STKs).
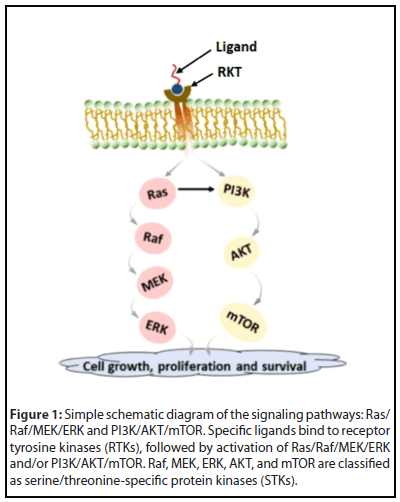
Mutation of Ras occurs in numerous malignancies, including pancreatic (90%), colon (45%), and lung (35%) [6,8]. Mammalian cells express three distinct but closely related Ras proteins (K-Ras, H-Ras, and N-Ras), which can become mutationally activated and, in turn, promote oncogenesis. The frequency of mutation of different Ras proteins varies, in which the K-Ras protein mutates most often [9].
The Ras-Raf-MEK-ERK signaling cascade can get activated by different stimuli (e.g., receptor tyrosine kinase and G-proteincoupled receptors). Mutations in Ras, as well as other upstream receptor genes, result in abnormal Ras-Raf-MEK-ERK signal activation. This specific pathway plays a significant role in the progress of hepatocellular carcinoma (HCC) and breast cancers [9]. Ras, Raf, MEK, ERK, and other associated molecules, are now considered therapeutic targets for cancer. Welsch et al. have described synthesis and testing of potential small-molecule pan-RAS ligands to interact with adjacent sites on the surface of oncogenic K-Ras. For example, the Ras-IN-3144 compound has shown binding and inhibiting the Ras signaling pathways in cancer cells and preventing the Ras mutant from growing in mouse cancer xenografts [10]. Sorafenib (Nexavar), a novel biaryl urea BAY 43-9006, is an orally administered multi-kinase inhibitor with activity against Ras/Raf kinases and several receptor tyrosine kinases such as vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), Fms-like tyrosine kinase-3 (FLT3), Ret, and c-Kit. It involves the angiogenic pathway and cell proliferation. Sorafenib has shown promising anti-tumor activity in in vitro studies, preclinical xenograft models of different tumor types, and human clinical trials [11-13]. PD184352 is an orally active and specific small-molecule inhibitor of one of the crucial components (MEK1/MEK2) of the MEK/ERK pathway and thereby effectively blocks the phosphorylation of ERK. The anti-tumor activity of this compound has passed in preclinical models, particularly for pancreas, colon, and breast cancers [14]. PD0325901 is also a specific MEK1/2 inhibitor and is a promising drug to treat thyroid cancers with either Ret/PTC or B-Raf mutation [15].
Wnt protein
Wnts are secreted factors that regulate cell growth, motility, and differentiation during embryonic development. Wnt signaling entails a multimolecular complex to control the intracellular levels of the dual functioning protein β-catenin [16,17]. This protein helps formation of cadherin adherens junction in epithelial cells and also activates genes that are associated with cell cycle progression.
Though there are two Wnt signaling pathways, namely, canonical (β-catenin dependent) and non-canonical (β-catenin independent). The canonical pathway is well-proven to be associated with cancer formation. The main intracellular proteins of this signaling pathway that control β-catenin levels are dishevelled (Dsh), axin-1, β-catenin, glycogen synthase kinase 3 (GSK-3), casein kinase 1α (CK-1α), and adenomatous polyposis coli (APC). A review article has elaborately described the canonical Wnt/β-catenin signaling pathway [18].
In the absence of Wnt ligands, β-catenin (β-cat) is phosphorylated by GSK3, which leads to β-cat degradation via β-TrCP200 ubiquitination and inhibits translocation of β-cat from the cytoplasm to the nucleus. Binding of Wnt to cell surface receptors Frizzled (Fz) and lipoprotein receptor-related protein-5/6 (LRP-5/6) triggers dismantling of the cytosolic β-cat destruction complex by associating the protein Dsh for membrane docking of Axin-1 and associated kinases, effecting the reduced degradation of β-cat and its accumulation in the nucleus [3]. Figure 2 shows a schematic diagram of the Wnt signaling pathway.
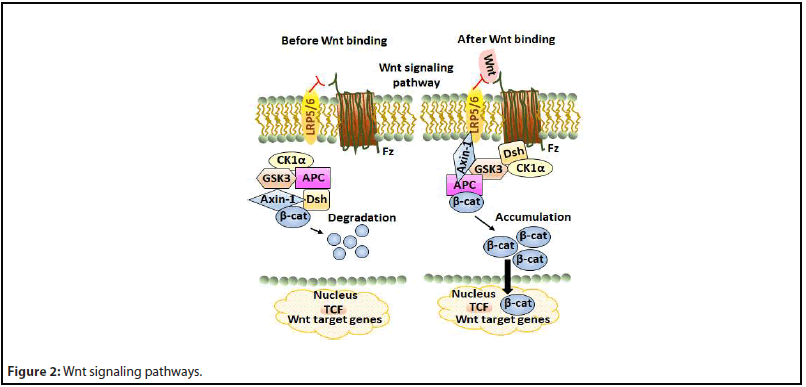
Aberrant Wnt/β-cat signaling is widely implicated in several cancers such as invasive ductal breast carcinomas [19], colorectal cancer [20,21], papillary thyroid cancer [22], esophageal cancer [23], leukemia, and melanoma [24]. Dysregulation of this signaling is commonly associated with mutations of Axin, APC, and β-cat leading to constitutive hyperactivation of this pathway [25]. Most colon cancers possess mutated APC genes, which eventually cause cancer [26,27]. But APC inactivation is not the only cause of colorectal cancer. About 92% of sporadic colorectal cancers involve at least one alteration in a Wnt pathway regulator [28]. Activation of the Wnt pathway in several breast cancers suggests that targeting components in this pathway could be a promising therapeutic strategy [29]. High levels of nuclear β-cat appear in several breast cancer subtypes in which canonical Wnt receptors and ligands overexpress. Anomalous Wnt signaling is a prime factor in leukemogenesis. In murine models of T-acute lymphoblastic leukemia (ALL), the expression of activated β-cat causes thymic lymphoma. Further, research suggests that β-cat is likely essential for the conversion of the pre-leukemia initiating cells (pre-LICs) to the LIC state, as well as for the self-renewal of LICs [30]. The role of the canonical Wnt signaling in melanoma is well established.
Numerous Wnt/β-cat pathway inhibitors have been examined in preclinical and clinical evaluations for different types of cancer, like colorectal, melanoma, and breast [6,31]. However, there are significant challenges in targeting the Wnt pathway; for example, finding efficacious agents without disturbing the normal somatic stem cell function in cellular repair and tissue homeostasis [24]. Porcupine (PORCN) is a membrane-bound O-acyltransferase required for palmitoylation of Wnt ligands, which is a necessary step in processing of the Wnt ligand secretion. Jang et al. investigated the effect and mechanism of the action of LGK974, a specific small-molecule PORCN inhibitor, in mice with lipopolysaccharide (LPS)-induced endotoxemia, an animal model of sepsis [32]. This study has revealed that LGK974 treatment has significantly and dosedependently increased the survival rate and reduced the plasma cytokine levels in mice with LPS-induced endotoxemia due to significant suppression in the expression of genes associated with Wnt/β-cat pathways, as well as with cytokine and NF-κB signaling. Another study has shown that LGK974 could inhibit proliferation and colony formation, and could induce apoptosis in clear cell renal cell carcinoma (ccRCC) cells by targeting the PORCN [33]. ETC-159 is another PORCN inhibitor. In preclinical studies, ETC-159 could induce tumor regression in patient-derived xenograft models of Wntaddicted cancers, including RNF43-mutant pancreatic cancers [34]. Another inhibitor is an anti-frizzled monoclonal antibody vantictumab (OMP-18R5) that binds to the Fz receptor and inhibits the canonical Wnt signaling [35].
The NF2 gene
The neurofibromatosis type-2 (NF2) gene encodes a cytoskeletal protein, called moesin-ezrin-radixin-like protein or merlin, also called Schwannomin. This protein forms in the nervous system, particularly in the specialized cells, called Schwann cells that wrap around axons of motor and sensory neurons to form the myelin sheath. Merlin helps regulate several signaling pathways that control the shape, growth, and adhesion of cells. Merlin also acts as a tumor suppressor that prevents cells from uncontrolled dividing [36].
As a cytoplasmic scaffolding protein, NF2 provides a crucial link between the extracellular environment and cell signaling pathways to prevent the effects of aberrant mitogenic signaling on oncogenic transformation [37,38]. Mutations and inactivation of the somatic NF2 may cause different cancers, such as Glioblastoma multiforme, breast, colorectal, skin, hepatic, and prostate cancers [36,39]. Alteration of the NF2 gene, which regulates the Hippo signaling pathway, seeds in 22.5% of papillary renal cell carcinomas (PRCC). The Hippo signaling pathway, shown in Figure 3, controls cell proliferation by regulating the transcriptional activity of the yes-associated protein (YAP). The loss of NF2 results in an aberrant YAP. Sourbier et al. have reported the potential inhibitory actions of dasatinib and saracatinib on aberrant YAP, leading to downregulation of YAP transcription targets, reduced cell viability, and G0-G1 cell cycle arrest. Xenograft models for NF2-deficient PRCC have also demonstrated a reduced tumor growth in the action of dasatinib [40].
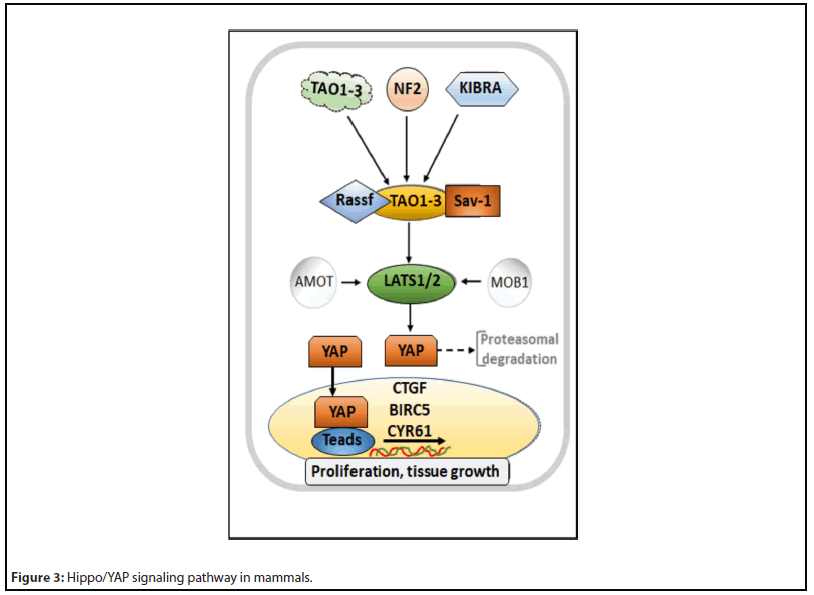
Hippo signaling pathway has been described elsewhere [41]. It involves several proteins, like MST1/2 protein kinases (mammalian STE20-like kinase 1/2) that get phosphorylated in their active states by NF2, KIBRA, or TAO1-3. Then there are Sav1 (Salvador) and Rassf (Ras association domain family) proteins, which, in association with MST1/2, activates large tumor suppressor-1/2 (LATS1/2) proteins. LATS1/2 phosphorylates YAP protein, which either retains in cytoplasm or degrades by proteasome. MOB1 (monopolar spindle-onebinder) and AMOT (angiomantin) proteins help in LATS1/2 phosphorylation and activities. When the Hippo signaling pathway is inactive, YAP is not phosphorylated, rather translocates in the nucleus to carry out transcriptional activity by binding to the TEAD (transcriptional enhanced associate domain). YAP, thus, regulates the expression of specific targets such as CTGF (connective tissue growth factor), BIRC5 (baculoviral inhibitor of apoptosis repeat-containing 5), or Cyr61 (cysteine-rich angiogenic inducer 61).
Signaling pathways in cancer stem cells
It is now believed that cancer stem cells (CSCs) develop tumors, though a tumor contains only 0.01–2% of CSCs [42]. CSCs have self-renewal capacity and differentiation potential, and contribute to several tumor malignancies, such as recurrence, metastasis, heterogeneity, and multidrug/ radiation resistance. Besides other factors, intracellular signaling pathways, such as Wnt, NF-κB (nuclear factor-κB), Hedgehog, and PI3K/AKT/mTOR, are the prime regulators of CSCs [43,44]. Here, we have briefly discussed Wnt and PI3K/ AKT/mTOR signaling pathways in CSCs.
Wnt signaling pathway: Stem cells and transcriptional circuits are related to the alteration or reactivation of several signaling pathways [45]. Turning on the Wnt pathway activates dormant CSCs that assist cell cycle progression using β-catenin [46]. The extracellular matrix (ECM) protein tenascin-C supports cell cycle in breast cancer cells by increasing Wnt signals [47]. Pyruvate kinase isozyme-M2 (PKM2) also plays an important role in the proliferation of breast CSCs [48-50]. An aberrant Wnt signal triggers the self-renewal of CSCs. Wnt/ β-cat pathway supports CSCs-mediated metastasis [51]. In cytomembrane, Fz promotes bone metastasis in prostate CSCs [52]. Long non-coding (lnc) RNAs and microRNAs promote self-renewal of CSCs through Wnt signaling pathway. LncTCF7 involves the SWI/SNF chromatin-remodeling complex to regulate the expression of TCF7 promoter in liver CSCs [53]. A small-molecule inhibitor CWP232228 has been reported to antagonize the binding of β-catenin to TCF in the nucleus to induce apoptosis in liver CSCs [54].
PI3K/AKT/mTOR signaling pathway: Phosphatasetensin homolog (PTEN) is a phosphatase in humans, which is encoded by the PTEN gene [55]. PTEN acts as a tumor suppressor gene through the action of its phosphatase protein product that regulates the cell cycle by preventing cells from growing and dividing too rapidly [56]. Studies show that mutations in PTEN lead to the inhibition of PI3K/mTOR signaling in glioblastoma multiforme. Deletion of PTEN in neural stem cells leads to a neoplastic phenotype such as cell growth promotion, resistance to cell apoptosis, and increase of migratory and invasive properties in vivo [57]. Activation of PI3K/Akt/mTOR signaling pathway enhances migration and invasion of prostate and pancreatic CSCs [58,59]. Activation of mTOR causes survival and proliferation of several CSCs such as breast and nasopharyngeal carcinoma CSCs [60,61], and colorectal CSCs [62].
Usually, signaling pathways are interconnected. Many such interlinked signaling channels are involved in CSCs. For example, the interconnection between the Notch and Wnt signaling pathways maintains an undifferentiated intestinal CSC [63]. A report has indicated that the spontaneous development of skin tumors in murine epidermis was due to activation of the Hedgehog and Wnt pathways [64].
Despite difficulty in finding a true efficacious drug, there are numerous success stories. For example, Imatinib (Gleevec) is the first oncogene-targeted therapy for cancer treatment. Imatinib mesylate, also known as STI571 or CGP57148, is a competitive inhibitor of a few tyrosine kinases, including BCRABL, ABL, KIT, and the platelet-derived growth factor receptors (PDGFR) [65]. It binds to the ATP-binding site of the target kinase and prevents phosphate transfer from ATP to tyrosine residues of various substrates and, thus, controls proliferative signaling in myeloid cells [66]. BCR-ABL oncogene is activated by translocating in chronic myelogenous leukemia (CML), and it is the driving force of leukemogenesis in CML [67,68]. Being a BCR-ABL inhibitor, imatinib is extremely efficacious in CSCs of CML [69,70].
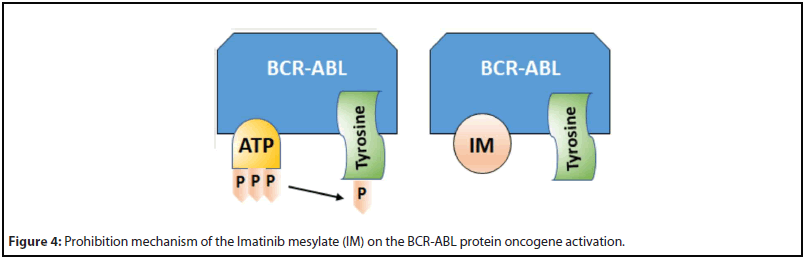
Figure 4 illustrates the prohibition mechanism of Imatinib mesylate (IM) upon activation of the BCR-ABL protein oncogene [71]. Normally, ATP binds to a specific pocket within the kinase domain of BCR-ABL protein and phosphorylates tyrosine residues on substrate proteins, as shown in the left in Figure 4. As IM has structural similarity with ATP, it can bind to the same ATP-binding pocket of BCR-ABL, as shown in the right in Figure 4. But IM carries no phosphate groups and thus inhibits the phosphorylation of tyrosine residues of the BCR-ABL protein, as described above. This way, IM prevents triggering of the downstream signaling pathway.
B-Raf inhibitors present another example that are extremely efficacious in B-Raf mutated melanoma. Several small-molecule B-Raf inhibitors have been developed, which have shown promising results in clinical trials for metastatic melanoma; vemurafenib and dabrafenib are two such inhibitors that have been clinically approved [72]. Vemurafenib, dabrafenib, and encorafenib have shown selective targeting to B-Raf kinase and interfering with the mitogen-activated protein kinase (MAPK) signaling pathway that regulates the proliferation and survival of melanoma cells [73]. Other examples include drugs targeting the estrogen receptor (ER) in breast cancer [74,75] and androgen receptor (AR) in prostate cancer [76-78], as well as monoclonal antibodies targeting the ErbB2 tyrosine kinase, such as trastuzumab, which has demonstrated efficacy, in particular, breast cancers (those that involve amplification of ErbB2 that drives uncontrolled mitogenic signaling) [79-81].
Nanomaterials to Target Aberrant Signaling Pathways in Cancer Cells
Though a growing number of drug inhibitors that target specific components of the signal pathways are in clinical use, the success of these agents has limitations due to resistance to the inhibitors in cancer cells [82]. Application of nanomaterials, the carriers of conventional drugs or therapeutic agents, to target aberrant signaling pathways in cancer therapy could be a better strategy to conquer this downside. Researchers are working in this direction.
In last few decades, clinical success in cancer therapeutics using kinase inhibitors has been around 5% only [83]. Besides the resistance to the targeting drugs, the drug-related toxicity, which arises due to off-target accumulation in the body, creates a serious barrier to an otherwise potent therapy. Exploring a nanomaterial-based approach for selectively delivering the inhibitor agents to cancer cells, while sparing healthy cells, may help to improve the efficacy, bioavailability, solubility, and stability and minimize the unwanted side effects [84,85].
Besides their small size and flexibility to compositional and surface modifications, nanoparticles can passively target solid tumors due to the enhanced permeability and retention (EPR) effect [86,87]. However, physiological barriers such as tumor penetration and heterogeneity, relative hypoxia, and endosomal escape may affect EPR effectiveness, necessitating the development of active targeting strategies [88]. The nanoparticles, surface-modified with different ligands such as antibodies, aptamers, peptides, or small molecules, can recognize tumor-specific antigens to actively target and bind at the tumor site [89]. The uptake of these nanoparticulates by the cancer cells becomes important in targeting aberrant cell signals. A cell can uptake nanoparticles through various mechanisms, such as phagocytosis, macropinocytosis, clathrindependent endocytosis, caveolin-dependent endocytosis, or crossing of membranes by diffusion depending on the size, shape, and surface character of the nanomaterials [90-92]. Usually, the internalization of nanoparticles follows an endocytosis mechanism.
Unfortunately, nanoparticles get covered with a layer of serum proteins or other biomolecules while passaging through body fluids and, thus, deviate from required activities like targeting specific cell membrane. Therefore, the challenge lies in limiting the interactions of nanoparticles with biomolecules or the immune system. It is achieved by functionalizing nanoparticles with inert polymers such as polyethyleneglycol (PEG), and/or smaller antibody fragments (e.g., scFv, Fab, F(ab’)2), or other homing molecules (e.g., aptamers, natural ligands), as well as self-markers [93]. Once successfully targeted and internalized, the drug-loaded nanoparticles can put off or demolish the faulty signals within the cancer cells to stop their uncontrolled proliferation, or promote expiry through apoptosis or necrosis. In the following, some research outcomes in this approach have been reviewed.
Ras pathway
Recently, Basu et al. have reported targeting of MAPK inhibitor PD98059, chemically conjugated with PLGA polymeric nanoparticles, to the MEK signaling pathway resulting in an enhanced antitumor efficacy of the cisplatin chemotherapy in melanoma tumor-bearing mice [94]. As reported by them, nanoparticles were taken up by cancer cells via endocytosis, wherein the active agent was released in a controlled manner resulting in inhibition of the phosphorylation of the downstream extracellular signal-regulated kinase. In another study, they encapsulated LY294002, a PI3K inhibitor, into PLGA nanoparticles and successfully targeted to inhibit the downstream AKT phosphorylation, resulting in inhibition of proliferation and apoptosis of B16/F10 melanoma cells in vitro [95]. The same drug-NPs composition could inhibit both B16/ F10- and MDA-MB-231-induced angiogenesis in an in vivo study with a zebrafish tumor xenograft model [95].
Selenium (Se) nanoparticles have several utilities in anticancer, immunoregulation, and drug-carrying activities. Pi et al. prepared oridonin-loaded and GE11 peptide conjugated Se nanoparticles that have shown an enhanced cellular uptake and, subsequently, cancer cell apoptosis by several processes, including activating the mitochondria-dependent pathway and inhibiting both EGFR/PI3K/AKT and Ras/Raf/MEK/ERK pathways [96]. GE11-conjugated Se NPs could actively target tumor tissues in an esophageal cancer-bearing mice. In a nude mice xenograft model, GE11-Ori-conjugated Se NPs could significantly inhibit tumor growth via inhibition of the tumor angiogenesis. In doing so, the NP-conjugates had reduced the expression of the angiogenesis-marker CD31 and activated the immune system by enhancing the production of both IL-2 and TNF-α [96].
Small interfering RNAs (siRNA) are clinically potential molecules for their precise regulation of gene expression. However, the effective delivery of RNAs to solid tumors is challenging. Recently, RNA interference targeting Ras has emerged [97]. Systemic delivery of SiRNAs by nanoparticles, nanoliposomes, or exosomes have exhibited anti-proliferative effects in cells by suppressing a mouse tumor [98-101]. Xue et al. have reported a successful delivery of P53-regulated miRNA mimics miR-34a to lung adenocarcinoma cells in vitro and to tumors in a genetically engineered mouse model with lung cancer, using SiRNAs-modified lipid/polymer nanoparticles and by targeting Kirsten rat sarcoma viral oncogene homolog (siKras). The outcome has revealed an impeded tumor growth due to reduced K-Ras gene expression and MAPK signaling, and increased apoptosis of cancer cells. In another example, novel hybrid nanoparticles, composed of human IgG and poloxamer-188, had escaped the clearance of macrophage, and delivered mutated K-Ras siRNA to A549 cells leading to an efficient knockdown of a mutated siRNA while protecting the siRNA from serum nuclease [100]. Strand et al. have reported the ability of a peptide-based, oligonucleotide condensing, endosomatic nanoparticle (NP) system to deliver siRNA to K-Ras-driven cancers [102]. They have shown that this peptidebased NP was taken up by cancer cells in vitro, wherein it delivered K-Ras-specific siRNA that has inhibited the K-Ras expression and reduced the cell viability.
Hyperactivated Ras regulates many oncogenic pathways in several human cancers, including glioblastoma, the most infiltrating, aggressive, and poorly treated brain tumor. Huang et al. synthesized apolipoprotein E3-reconstituted high-density lipoprotein (ApoE-rHDL) encapsulated calcium phosphate (CaP) nanoparticle core, named as CaP-rHDL [103], for tumor-targeting siRNA delivery, activating transcription factor-5 (ATF5), an overexpressed anti-apoptotic transcription factor in glioblastoma [104,105]. They investigated in vivo tumor-targeting efficiency of CaP-rHDL and its cellular uptake through micropinocytosis in intracranial C6 and GICs glioblastoma-bearing mice models, and observed an efficient delivery of ATF5 siRNA to Ras-activated brain cancer cells [103].
Dysregulation of mTOR signaling in cancer makes it an attractive therapeutic target, and, thus, several types of mTOR inhibitors have been developed. But their clinical acceptance has been hampered by their pharmacokinetic limitations. Nanoparticle-based systems can overcome these unfathomable biological barriers and release their therapeutic loads at the targeted sites. Recently, Yoon has reviewed such a possibility using the molecular mechanism of nanoparticlebased mTOR modulator action [106]. Cancer cell lysosomes play a critical role in amino acid-induced mTORC1 activation [107]. Most nanoparticles accumulate in the acidic vesicular organelles, containing hydrolytic enzymes such as endosomes and lysosomes [108,109], and degrade [106]. Therefore, targeting lysosomes using nanoparticles may present an effective strategy for anticancer therapy via regulation of the mTOR signaling [110-112]. For example, nano-SiO2 blocks the PI3K/Akt/mTOR pathway, leading to an endothelial cell dysfunction [110]. However, modulation of the mTOR action by nanoparticles has low specificity. Therefore, conjugation of selective mTOR inhibitors with nanoparticles could increase their therapeutic effects [113]. More research on nanoparticlebased inhibition of the Ras-mediated signaling pathways in different cancers will be helpful.
Wnt pathway
Wnt pathway seems to be a promising target for therapeutic needs. Thus, antibodies and chemo-therapeutic reagents have been developed to target aberrant Wnt/β-catenin signaling pathways [114-122]. But their pharmacological inhibition faces challenges owing to substantial complexity within the track and the interlink between different pathways. Silica (SiO2) NPs with optimum size, surface charge, porosity, and dosage are minimally cytotoxic and biocompatible; therefore, attractive for biomedical imaging and therapeutic applications [123,124]. However, Yi et al. have shown that unmodified SiO2 NPs could actively affect Wnt signaling in various cancer cell lines and impaired the Wnt-regulated embryonic development in Zebrafish [125]. They have also demonstrated that intracellular SiO2 NPs could block Wnt signal transduction by targeting the Dsh protein for lysosomal degradation.
Valcourt et al. have reviewed the targeting approaches to different signaling pathways, like Wnt pathway, using nanoparticles [126]. For example, recombinant secreted frizzled-related protein-1 (SFRP1)-bound luminescent gold nanocluster embedded nanoparticles have been used to target the canonical Wnt signaling pathway in the microenvironment of cervical cancer cells, which showed anti-proliferative effects by limiting the expression levels of Wnt target proteins, i.e., β-catenin, cyclin D1, and survivin [127]. SFRP1 is a naturally produced Wnt inhibitor. It blocks Wnt signaling by binding to extracellular Wnt ligands, forbidding them from binding to Wnt receptors [128,129], or directly binding to the Fz receptors [128]. Liu et al. used cationic lipid-protamine nanoparticles to trap Wnt5a molecules in cancer cells to treat metastatic melanoma by preventing the signaling feedback [130]. The NP system could deliver plasmid DNA, encoding a trimeric trap protein consisting of an extracellular domain of Fz receptors, to tumors. Consequently, tumors had expressed trap-protein to bind Wnt5a and reduce its intratumoral expression, and, thus, limiting the tumor growth, particularly when combined with doxorubicin chemotherapy [130]. Besides natural Wnt inhibitors, synthetic inhibitors such as Niclosamide [131] and cromolyn [132] have also been incorporated into NP carriers for anti-cancer therapy.
Above approaches lack targetability to the primary mediator of the Wnt pathway, β-catenin, because it lacks an effective binding site for small molecule therapeutics [126]. An alternate way to suppress β-catenin and other significant proteins in the Wnt pathway is to use siRNA or miRNA for RNA interference (RNAi) mediated gene silencing. It has been achieved using lipid nanoparticles (LNPs) containing Dicer substrate siRNA (DsiRNA) targeting CTNNB1, the gene encoding β-catenin, which has achieved inhibition of tumor growth in Wntdependent colorectal and hepatocellular carcinoma models in mice [133]. Researchers have developed other complexes as well for exploring RNAi-mediated gene silencing; for example, polyethylene glycol-polyethyleneimine-chlorin-e6 (PEG-PEICe6) NPs could deliver Wnt-1 siRNA to oral cancer cells and enable simultaneous photodynamic therapy (PDT), chlorin e6 being a photosensitizer [134]. Often PDT is curbed by the Wnt signal-mediated epithelial-to-mesenchymal transition. Thus, silencing of Wnt-1 helps PDT.
A small molecule or RNA-based therapy works inside the cell, whereas antagonistic antibodies can work outside the cell. An antagonistic antibody typically binds its target receptor in the cell membrane and makes it ligand-unresponsive and, thus, hinders respective signaling pathway within the cell. Recently, researches have revealed that antibody-conjugated nanoparticles are more effective for this approach than free antibodies [135,136]. Peptide-conjugated nanoparticles can also act like antibody-nanoparticle conjugates, as demonstrated by Miller-Kleinhenz et al. using peptide-coated ultra-small magnetic iron oxide nanoparticles (IONPs) to target both Wnt/LRP5/6 and uPA receptors [137]. This dualtargeting successfully inhibited the Wnt/β-cat signaling pathways in breast cancer cells by reducing the expression of β-catenin, Axin, GSK-3β, and Snail [137]. Further exploration of nanoparticle-based systems for the inhibition of Wnt signaling pathways in different cancer cells is needed.
Protein-Nanoparticle Interactions and Signaling Pathways in Cancer cells
Protein-nanoparticle interaction plays a crucial part in biomedical applications of nanoparticles [138-144]. This interaction starts as soon as nanoparticles enter the physiological medium and get coated with a layer of biomolecules/proteins, called ‘corona’ [145-148]. Many factors such as size, shape, and surface charge of nanoparticles, pH of the medium, and the size and conformation of the biomolecules/proteins influence this interaction [149,150]. Huang et al. have reported the effects of surface compositional and structural heterogeneity on protein adsorption by examining the interaction of self-assembled monolayer coated gold NPs (AuNPs) with two types of proteins: ubiquitin and fibrinogen [150]. The surface coating on nanoparticles with charged and neutral molecules shows different affinities to the binding of proteins and it causes irreversible conformational unfolding of proteins if counterions are present in the coating molecules [151-156]. Corona alters the size and interfacial composition of a nanoparticle, giving it a distinct ‘biological identity’ from that of the original one [157,158]. This new identity determines the physiological responses of nanomaterials to various activities like cell signaling, kinetics, transport, accumulation, and toxicity in vivo [158]. Though a suitable surface coating such as PEGylation can reduce the binding of biomolecules/proteins [154,159], some association yet may occur [160,161]. In this section, we shall discuss the effect of the biological identity of nanoparticles on cellular uptake and signaling pathways.
With matching size and surface, nanoparticle-corona composites participate in a wide range of endogenous cellular uptake and other processes [160,162-164], interact with every cellular and organ component, and initiate cell signaling [165,166]. The recognition of protein corona by the biological machinery leads to specific interactions between them, resulting in more specifically regulated pathways [156]. Both composition and conformation of the absorbed biomolecules determine the impacts of the biomolecule-nanoparticle complexes [165,167-173]. For instance, nanoparticle-induced protein unfolding had initiated the nuclear factor-κB (NF-κB) pathway and inflammation in the cell [165].
Hussain et al. have reviewed nanomaterial-induced oxidative stress-dependent and independent signaling pathways, epigenetic regulation by nanomaterials, and the effects of the nanoparticle-protein interactions on cell signaling pathways [174]. They pointed out that activation or inactivation of receptor-dependent cell signals controls the biological efficacies of nanomaterials. For example, Rosso et al. have reported that plasma vitronectin-bound nanomaterials (e.g., maleic anhydride/alkyl vinyl ethers-based NPs) triggered the activation of the vitronectin-integrin receptor leading to increased phosphorylation of ERK1/2 and FAK kinases that had increased proliferation and cell cycle progression [174]. On the other hand, nanoparticle-induced oxidative stress can activate a wide variety of cellular events such as cell cycle arrest, apoptosis, inflammation, and induction of antioxidant enzymes upon activation of cellular pathways [175]. The effect of surface charge and counterions of coated nanoparticle surfaces can induce necrotic cell death in cancer cells without affecting any normal cells, as observed in an in vitro experiment [176].
The success of the nanoparticle-based drug delivery for inhibition of any aberrant cell signaling depends on the level and mechanism of the cellular uptake of drug-nanoparticle conjugates; biomolecular/protein corona on nanoparticles plays a pivotal role in this. Oh et al. have reported that gold (Au) NP-protein complexes could be recognized by the membrane receptors and internalized by the cells [177]. The formation of the corona and conformation of the adsorbed biomolecules/proteins on nanoparticles depend on various factors such as size and shape of nanoparticles and their surface chemistry [178]. Au NPs can be internalized by the cells by receptor-mediated endocytosis and phagocytosis pathways. According to a report, both these pathways got influenced by the following factors: corona on Au NPs, recognition by the membrane receptors; engulfment of the complexes by the cytosolic vesicles being transported or penetrated in cells; sequential trafficking inside the cells; and storage or elimination of Au NPs by the cells [179].
The applicability of nanoparticles in various biological activities critically depends on their surface functionalization and biodegradability. Lunova et al. have demonstrated that amino-functionalized polystyrene nanoparticles (PS-NH2) could trigger cell death in hepatocellular carcinoma Huh7 cells, which was not possible with amino- or hydroxyl-functionalized silica particles [180]. They explained it by considering molecular level interactions; for example, PS-NH2 obstructed, and aminofunctionalized silica nanoparticles (Si-NH2) activated the mTOR signaling in Huh7 and HepG2 cells. The PS-NH2 induced timedependent lysosomal destabilization was associated with the damage that had occurred in the mitochondrial membrane. In PS-NH2-treated cells, permeabilization of lysosomes was the cause of cell death. In contrast, Si-NH2 nanoparticles had enhanced the proliferation of HuH7 and HepG2 cells [180]. Several other groups also have reported the modulation of the mTOR pathway by NPs, resulting in cell-cycle arrest in leukemia cells [111,112,181,182].
Translating nanoparticle-based agents from laboratory into clinical applications faces a challenge due to the difficulty in eliminating interactions on the interfaces between surface-engineered nanoparticles and biological systems. As mentioned earlier, PEGylation can only reduce the binding of biomolecules/proteins on the nanosurface [159]. Kang et al. have reported mannose-modified PEGylated hydroxyethyl starch nanocarriers (HES-NCs) to target dendritic cells (DCs) [183]. In this case, the interaction of nanoparticle composite with human plasma had caused a low overall protein binding and a high specific affinity to DCs binding. However, these strategies are limited at conferring the targeting specificity [184-190]. Another group has reported a more robust approach to minimize the interaction of nanoparticles with serum proteins [191]. They used a recombinant fusion protein, GST-HER2-Afb, as a protein corona shield, in which HER2-binding affibody (Afb) was genetically combined with a glutathione-S-transferase (GST), a well-known fusion tag protein, with an extra linker that could reduce serum protein absorption while retaining its targeting specificity [191]. Further research may help to achieve in vivo targeting of functionalized nanoparticles to aberrant signaling pathways in cancer cells.
Conclusions and Future Proposal
Based on various reports cited in this article, we conclude that more robust and easier strategies for nanoparticle functionalization must evolve that would ensure specific in vivo targeting of nanocarriers for the delivery of therapeutic drugs to inhibit aberrant signaling pathways in cancer cells. Targeted delivery can minimize the side effects of drugs in healthy cells and reduce the drug requirement in the body. Alternately, we propose the following strategies of drugless therapy.
Ion-based therapy
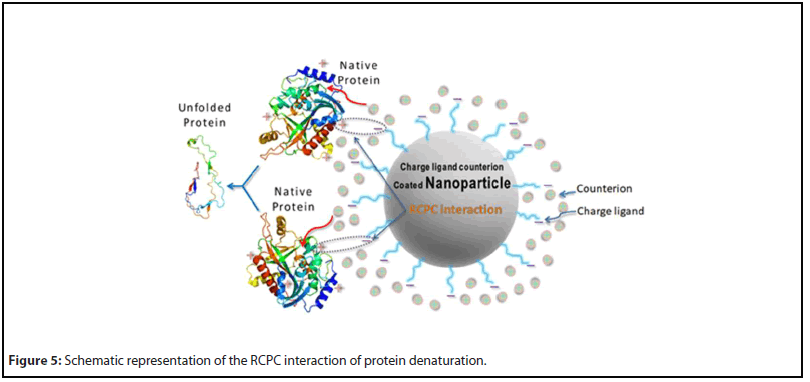
Earlier, Ghosh et al. have proposed an interaction model, reverse-charge-parity counterions (RCPC) interaction, between biomolecules and counterion-conjugated charged nanoparticles that had caused irreversible denaturation of proteins [152,153]. The same interaction model has been reported to cause a necrotic death of cancer cells by disintegrating their cell membranes in an in vitro experiment [176]. On the other hand, in vivo experiments face challenges due to formation of biomolecular corona that gives the precoated nanoparticles a new ‘biological identity’ [145-148,156-158]. This new identity causes unpredictable outcomes such as losing the targetability and unregulated toxicity of the nanoparticles. To minimize the protein binding, several coatings on nanoparticles have been proposed [159,183,191]. In a recent review, Ghosh and Panicker have proposed a composite coating by PEGylation of the nanoparticle surface, grafting of counterion-conjugated charged molecules, and a coating of specific antibodies using suitable linker molecules [176]. The utility of this composite coating is the following. PEGylation can protect nanoparticles from serum protein and other biomolecular adsorption in the physiological medium, antibody can help targeting specific marker proteins in cell membranes, and counterion-conjugated charged molecules can perform the RCPC interaction with the proteins in cell membrane or in cytosolic medium of cancer cells. If specific transmembrane proteins such as receptor kinase tyrosine (RKT), responsible for the downstream cell signaling, can be targeted, the counterion-conjugated charged molecules on nanoparticles will initiate the RCPC interaction and denature receptor kinases and disrupt cell signaling. Besides, if nanoparticle-conjugates are internalized by the targeted cancer cells via endocytosis, they can bind to proteins in the signaling pathway and destroy them through the RCPC interaction. Work in this direction will be truly beneficial. Figure 5 shows the scheme of the RCPC interaction of protein denaturation upon interaction with ion-conjugated charged nanoparticles.
Photothermal therapy
Photothermal therapy (PTT) relies on nanoparticle-based light absorbers that convert light to localized heat energy for treating cancer, microbial, and other diseases [192,193]. PTT originated with gold nanostructures, such as nanorods, nanoshells, nanocages, and nanostars, that show size, shape and design-tunable plasmonic absorption in the visible-NIR region [194,195]. When such nanostructures accumulated in solid tumors following either systemic or intratumoral administration, subsequent light irradiation led to robust tumor ablation with minimal side effects. The inert properties of gold, combined with the ease of their surface modification with passivating polymers and/or biorecognition agents, facilitates their prolonged systemic circulation and tumor accumulation [196]. The mechanism of photothermal cell death is often necrosis, though other mechanisms such as apoptosis has also been reported [192]. In a recently concluded clinical pilot trial involving the laser-excited gold nanoshells for the treatment of prostate cancer in men, in combination with MRI-ultrasound fusion imaging, it has reported that the focused tumor ablation could be achieved in 94% of patients, without significant side-effects [197].
Subsequently, PTT has been found in several other nanostructures, such as doped semiconductors, magnetic nanomaterials, etc. [198,199]. Roy and colleagues have found that the well-known pigment Prussian Blue (PB), and its analogues (PBAs) can serve as efficient nanoparticulate photothermal agents [200]. They have demonstrated efficient bactericidal and anticancer action using a formulation of chitosan-coated PB nanoparticles irradiated with red-emitting laser light [201]. Recently, it has been recognized that localized PTT also triggers a systemic immunogenic response that affects cancer cells in non-target regions (abscopal effect) [192,202]. Therefore, combination of PTT with tumor immunotherapy, such as involving checkpoint inhibitors, are being actively explored for treating not only solid tumors, but also distant metastases to prevent tumor recurrence and facilitate longterm cancer therapy [203].
Magnetic hyperthermia therapy
Magnetic hyperthermia therapy (MHT) is another exciting drug-free anticancer therapy that involves tumor-ablation via localized heat generation (hyperthermia) resulting from the exposure of tumor-targeted magnetic nanoparticles to external AC magnetic field [204,205]. Several kinds of magnetic nanoparticles, such as iron oxides, ferrites, etc. can show this effect via a variety of mechanisms, that have been reviewed elsewhere [206]. The ultra-small size and facile surface chemistry of such nanoparticles allows their minimally impeded systemic circulation and tumor-homing tendency [207]. In addition, these nanoparticles can be arrested near tumor sites with the aid of externally applied magnetic force (magnetic targeting) [208]. Finally, like in other ablation therapies, the mechanism of the cell death involving MHT is primarily necrosis [205]. The magnetic-field activated aminosilane-coated superparamagnetic iron oxide nanoparticles, in combination with radiation therapy, has been applied for the clinical treatment of glioblastoma in humans. Enhanced overall survival in the treatment group was observed when compared to the non-treated (control) group, with no evidence of significant toxic side effects [209].
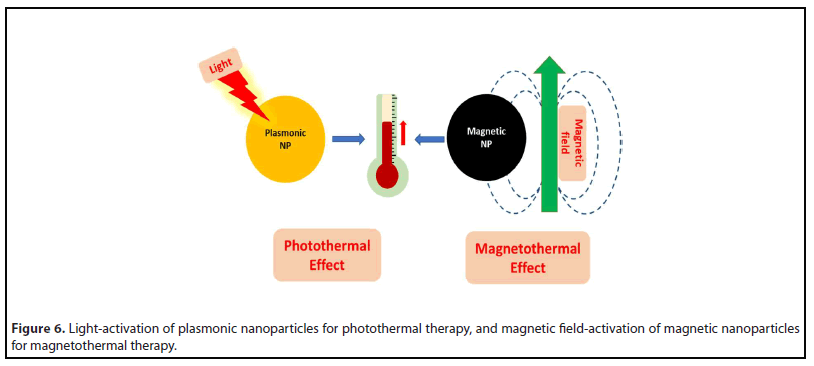
An advantage of iron-oxide nanoparticles is their ability to serve as contrast agents for magnetic resonance imaging (MRI) [207]. Other functionalities can also be incorporated with these nanoparticles for multimodal therapeutics and theragnostics. For example, Roy and his colleagues have used photosensitizer-doped cobalt ferrite nanoparticles for bimodal photo- and magnetic-field induced therapy of cancer in vitro [210]. Moreover, like in PTT, localized MHT is also known to trigger a systemic immune response, that can be exploited for combined MHT and immunotherapy of cancer [211]. Figure 6 shows the schemes of photothermal (PTT) and magnetic hyperthermia (MHT) therapies.
Author Contributions
IR and GG formulated the topic of the review. GG wrote the manuscript and prepared figures. IR critically revised the manuscript and the figures, and furnished additional information.
Declaration of Competing Interests
The authors declare that the preparation of this review article required no commercial or financial from any agency.
Acknowledgements
GG gratefully acknowledge Dr. Lata Panicker for fruitful collaborations on protein-nanoparticle interaction studies. He is also thankful to Dr. Rita Mukhapadhyaya and other collaborators in cancer cell research. IR acknowledges Institute for Eminence (IoE), University of Delhi, for providing funding support.
References
2. Lodish H, Berk A, Kaiser CA, Krieger M, Scott MP, Bretscher A, et al. Cell signaling I: Signal transduction and short-term cellular processes. Molecular Cell Biology, 6th ed., New York, WH Freeman and Company (2008) pp. 623–64; ISBN 978-0716776017.
3. Juliano RL. Addressing cancer signal transduction pathways with antisense and siRNA oligonucleotides. NAR Cancer. 2020 Sep;2(3):zcaa025.
4. Martin GS, Cell signaling and cancer. Cancer Cell. 2003 Sep;4(3):167-74.
5. Sever R, Brugge JS. Signal transduction in cancer. Cold Spring Harbor Perspectives in Medicine. 2015 Apr 1;5(4):a006098.
6. Prior IA, Lewis PD, Mattos C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Research. 2012 May 15;72(10):2457- 67.
7. Giancotti FG. Deregulation of cell signaling in cancer. FEBS Letters 2014 Aug 19;588(16):2558-70.
8. Simanshu DK, Nissley DV, McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell. 2017 Jun 29;170(1):17-33.
9. Li L, Zhao GD, Shi Z, Qi LL, Zhou LY, Fu ZX. The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC. Oncology Letters. 2016 Nov;12(5):3045-50.
10. Welsch ME, Kaplan A, Chambers JM, Stokes ME, Bos PH, Zask A, Zhang Y, et al. Multivalent Small Molecule pan-RAS Inhibitors. Cell. 2017 February 23;168(5):878-89.
11. Ranieri G, Gadaleta-Caldarola G, Goffredo V, Patruno R, Mangia A, Rizzo A, et al. Sorafenib (BAY 43-9006) in Hepatocellular Carcinoma Patients: From Discovery to Clinical Development. Current Medicinal Chemistry 2012;19(7):938-44.
12. Adnane L, Trail PA, Taylor I, Wilhelm SM. Sorafenib (BAY 43-9006, Nexavar®), a dual-action inhibitor that targets RAF/MEK/ ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods in Enzymology. 2006;407:597-612.
13. Voliotis D, Dumas J. Clinical Development of Sorafenib (BAY 43-9006) VEGFR and RAF Inhibitor. In: Marmé D., Fusenig N., eds, Tumor Angiogenesis. Springer, Berlin, Heidelberg (2008).
14. Allen LF, Sebolt-Leopold J, Meyer MB. CI-1040 (PD184352), a targeted signal transduction inhibitor of MEK (MAPKK). Seminars in Oncology 2003 Oct;30(5 Suppl 16):105-16.
15. Henderson YC, Chen Y, Frederick MJ, Lai SY, Clayman GL. MEK Inhibitor PD0325901 Significantly Reduces the Growth of Papillary Thyroid Carcinoma Cells In vitro and In vivo. Molecular Cancer Therapeutics 2010 July;9(7):1968-76.
16. van Kappel EC, Maurice MM. Molecular regulation and pharmacological targeting of the ß-catenin destruction complex. British Journal of Pharmacology. 2017 Dec;174(24):4575-88.
17. Nusse R, Clevers H. Wnt/ß-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell. 2017 Jun 1;169(6):985-99.
18. Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis. 2008 Apr;4(2):68-75.
19. Abd El-Rehim D, Ali MM. Aberrant expression of ß-catenin in invasive ductal breast carcinomas. Journal of the Egypt National Cancer Institute. 2009 Jun;21(2):185-95.
20. Adachi S, Jigami T, Yasui T, Nakano T, Ohwada S, Omori Y, et al. Role of a BCL9-Related ß-Catenin-Binding Protein, B9L, in Tumorigenesis Induced by Aberrant Activation of Wnt Signaling. Cancer Research 2004;64:8496-501.
21. Pan T, Xu J, Zhu Y. Self-renewal molecular mechanisms of colorectal cancer stem cells. International Journal of Molecular Medicine. 2017 Jan;39(1):9-20.
22. Ishigaki K, Namba H, Nakashima M, Nakayama T, Mitsutake N, Hayashi T, et al. Aberrant localization of beta-catenin correlates with overexpression of its target gene in human papillary thyroid cancer. The Journal of Clinical Endocrinology and Metabolism 2002 Jul;87(7):3433-40.
23. Kudo J, Nishiwaki T, Haruki N, Ishiguro H, Shibata Y, Terashita Y, et al. Aberrant nuclear localization of beta-catenin without genetic alterations in beta-catenin or Axin genes in esophageal cancer. World Journal of Surgical Oncology 2007 Feb 19;5:21.
24. Krishnamurthy N, Kurzrock R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treatment Reviews. 2018 Jan;62:50-60.
25. White BD, Chien AJ, Dawson DW. Dysregulation of Wnt/ß- catenin signaling in gastrointestinal cancers. Gastroenterology. 2012 Feb;142(2):219-32.
26. Novellasdemunt L, Antas P, Li VSW. Targeting Wnt signaling in colorectal cancer. A Review in the Theme: Cell Signaling: Proteins, Pathways and Mechanisms. American Journal of Physiology: Cell Physiology 2015 Oct 15;309(8):C511-21.
27. Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. Journal of Cell Science. 2007 Oct 1;120(19) 3327-35.
28. Schatoff EM, Leach BI, Dow LE. Wnt Signaling and Colorectal Cancer. Current Colorectal Cancer Reports. 2017 April;13(2):101-10.
29. Khramtsov AI, Khramtsova GF, Tretiakova M, Huo D, Olopade OI, Goss KH. Wnt/ß-Catenin Pathway Activation Is Enriched in Basal-Like Breast Cancers and Predicts Poor Outcome. The American Journal of Pathology. 2010 Jun;176(6):2911-20.
30. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017 Mar;36(11):1461-73.
31. Zhang Y, Wang X. Targeting the Wnt/ß-catenin signaling pathway in cancer. Journal of Hematology & Oncology. 2020 Dec;13(1):165.
32. Jang J, Song J, Lee H, Sim I, Kwon YV, Jho Eh. LGK974 suppresses lipopolysaccharide-induced endotoxemia in mice by modulating the crosstalk between the Wnt/ß-catenin and NF-κB pathways. Experimental & Molecular Medicine. 2021;53:407-21.
33. Li J, Wu G, Xu Y, Li J, Ruan N, Chen Y, et al. Porcupine Inhibitor LGK974 Downregulates the Wnt Signaling Pathway and Inhibits Clear Cell Renal Cell Carcinoma. BioMed Research International. 2020 Feb 13;2020:2527643.
34. Zhong Z, Sepramaniam S, Chew XH, Wood K, Lee MA, Madan B, et al. PORCN inhibition synergizes with PI3K/mTOR inhibition in Wnt-addicted cancers. Oncogene. 2019 Oct;38(40): 6662-77.
35. Diamond JR, Becerra C, Richards D, Mita A, Osborne C, O’Shaughnessy J, et al. Phase Ib clinical trial of the anti-frizzled antibody vantictumab (OMP-18R5) plus paclitaxel in patients with locally advanced or metastatic HER2-negative breast cancer. Breast Cancer Research and Treatment. 2020 Nov;184(1):53-62.
36. Cooper J, Giancotti FG. Molecular insights into NF2/ Merlin tumor suppressor function. FEBS Letters. 2014 Aug 19;588(16):2743-52.
37. Beltrami S, Kim R, Gordon J. Neurofibromatosis Type 2 Protein, NF2: An uncoventional cell cycle regulator. Anticancer Research. 2013 Jan;33(1):1-11.
38. Morrison H, Sherman LS, Legg J, Banine F, Isacke C, Haipek CA, et al. The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes & Development. 2001 Apr 15;15(8):968-80.
39. Petrilli AM, Fernández-Valle C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene. 2016 Feb 4;35(5):537-48.
40. Sourbier C, Liao PJ, Ricketts CJ, Wei D, Yang Y, Baranes SM, et al. Targeting loss of the Hippo signaling pathway in NF2-deficient papillary kidney cancers. Oncotarget. 2018 Jan 10;9(12):10723-33.
41. Morice S, Danieau G, Rédini F, Brounais-Le-Royer B, Verrecchia F. Hippo/YAP Signaling Pathway: A Promising Therapeutic Target in Bone Paediatric Cancers? Cancers. 2020 March 10;12(3):645.
42. Cojoc M, Mabert K, Muders MH, Dubrovska A. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms. Seminars in Cancer Biology 2015 Apr;31:16-27.
43. Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduction and Targeted Therapy. 2020 Feb 7;5(1):8.
44. Espinosa-Sánchez A, Suárez-Martínez E, Sánchez-Díaz L, Carnero A. Therapeutic Targeting of Signaling Pathways Related to Cancer Stemness. Frontiers in Oncology. 2020 Aug 26;10: 1533.
45. Clevers H, Nusse R, Wnt/ß-catenin signaling and disease. Cell. 2012 Jun 8;149(6):1192-205.
46. Giancotti FG, Mechanisms governing metastatic dormancy and reactivation. Cell. 2013 Nov 7;155(4):750-64.
47. O’Connell JT, Sugimoto H, Cooke VG, MacDonald BA, Mehta AI, LeBleu VS, et al. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proceedings of the National Academy of Sciences of the United States of America. 2011 Sep 20;108(38):16002-7.
48. Liang J, Cao R, Wang X, Zhang Y, Wang P, Gao H, et al. Mitochondrial PKM2 regulates oxidative stress-induced apoptosis by stabilizing Bcl2. Cell Research 2017 Mar;27(3):329-51.
49. Weinberg RA. The many faces of tumor dormancy. Acta Pathologica, Microbiologica, et Immunologica Scandinavica. 2008 Jul-Aug;116(7-8):548-51.
50. Zhao Z, Song Z, Liao Z, Liu Z, Sun H, Lei B, et al. PKM2 promotes stemness of breast cancer cell by through Wnt/ß-catenin pathway. Tumour Biology 2016 Mar;37(3):4223-34.
51. DiMeo TA, Anderson K, Phadke P, Fan C, Perou CM, Naber S, et al. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Research. 2009 Jul 1;69(13):5364- 73.
52. Li Q, Ye L, Zhang X, Wang M, Lin C, Huand S, et al. FZD8, a target of p53, promotes bone metastasis in prostate cancer by activating canonical Wnt/ß-catenin signaling. Cancer Letters. 2017 Aug 28;402:166-76.
53. Wang Y, He L, Du Y, Zhu P, Huang G, Luo Z, et al. The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell. 2015 Apr 2;16(4):413-25.
54. Kim JY, Lee HY, Park KY, Choi YK, Nam JS, Hong IS. CWP232228 targets liver cancer stem cells through Wnt/ß-catenin signaling: a novel therapeutic approach for liver cancer treatment. Oncotarget. 2016 Apr 12;7(15):20395-409.
55. Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers, Nature Genetics. 1997 Apr;15(4):356-62.
56. Chu EC, Tarnawski AS. PTEN regulatory functions in tumor suppression and cell biology. Medical Science Monitor. 2004 Oct;10(10):RA235-41.
57. Duan S, Yuan G, Liu X, Ren R, Li J, Zhang W, et al. PTEN deficiency reprogrammes human neural stem cells towards a glioblastoma stem cell-like phenotype. Nature Communications. 2015 Dec 3;6:10068.
58. Chang L, Graham PH, Hao J, Ni J, Bucci J, Cozzi PJ, et al. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/ mTOR pathway in prostate cancer radioresistance. Cell Death & Disease. 2013 Oct 24;4:e875.
59. Fitzgerald TL, Lertpiriyapong K, Cocco L, Martelli AM, Libra M, Candido S, et al. Roles of EGFR and KRAS and their downstream signaling pathways in pancreatic cancer and pancreatic cancer stem cells. Adv. Biol. Regul. 2015 Sep;59:65-81.
60. Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J, et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proceedings of the National Academy of Sciences of the United States of America. 2007 Oct 9;104(41):16158-63.
61. Yang C, Zhang Y, Zhang Y, Zhang Z, Peng J, Li Z, et al. Downregulation of cancer stem cell properties via mTOR signaling pathway inhibition by rapamycin in nasopharyngeal carcinoma. International Journal of Oncology. 2015 Sep;47(3):909-17.
62. Huang EH, Hynes MJ, Zhang T, Ginestier C, Dontu G, Appelman H, et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Research. 2009 Apr 15;69(8):3382-9.
63. van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas intogoblet cells. Nature. 2005 Jun 16;435(7044):959-63.
64. Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, et al. Notch1 functions as a tumor suppressor in mouse skin. Nature Genetics. 2003 Mar;33(3):416-21.
65. Iqbal N, Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. Chemotherapy Research and Practice. 2014 May 19;2014:357027.
66. Joensuu H, Dimitrijevic S. Tyrosine kinase inhibitor imatinib (STI571) as an anticancer agent for solid tumours. Annals of Medicine. 2001 Oct;33(7):451-5.
67. Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science 1990 Mar 2;247(4946):1079-82.
68. Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. The New England Journal of Medicine. 2001 Apr 5;344(14):1038-42.
69. Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature Medicine. 1996 May;2(5):561-6.
70. Chen Y, Peng C, Sullivan C, Li D, Li S. Novel therapeutic agents against cancer stem cells of chronic myeloid leukemia. Anti-Cancer Agents in Medical Chemistry. 2010 Feb;10(2):111-5.
71. Vandenberghe P, Boeckx N, Ronsyn E, Decorte R, Verhoef G, Hagemeijer A. Imatinib mesylate induces durable complete remission of advanced CML persisting after allogeneic bone marrow transplantation. Leukemia. 2003 Feb;17(2):458-60.
72. Johansson CH, Brage SE. BRAF inhibitors in cancer therapy. Pharmacology & Therapeutics. 2014 May;142(2):176-82.
73. Proietti I, Skroza N, Michelini S, Mambrin A, Balduzzi V, Bernardini N, et al. BRAF Inhibitors: Molecular Targeting and Immunomodulatory Actions. Cancers (Basel). 2020 Jul 7;12(7):1823.
74. Ariazi EA, Ariazi JL, Cordera F, Jordan VC. Estrogen receptors as therapeutic targets in breast cancer. Current Topics in Medicinal Chemistry. 2006;6(3):181-202.
75. Shanle EK, Xu W. Selectively targeting estrogen receptors for cancer treatment. Advanced Drug Delivery Reviews. 2010 Oct 30;62(13):1265-76.
76. Fujita K, Nonomura N. Role of Androgen Receptor in Prostate Cancer: A Review. The World Journal Men’s Health. 2019 Sep;37(3):288-95.
77. Tan MHE, Li J, Xu HE, Melcher K, Yong El. Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacologica Sinica. 2015 Jan;36(1):3-23.
78. Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocrine Reviews. 2004 Apr;25(2):276-308.
79. Nielsen DL, Andersson M, Kamby C. HER2-targeted therapy in breast cancer. Monoclonal antibodies and tyrosine kinase inhibitors. Cancer Treatment Reviews. 2009 Apr;35(2):121-36.
80. Nielsen DL, Kümler I, Palshof JAE, Andersson M. Efficacy of HER2-targeted therapy in metastatic breast cancer. Monoclonal antibodies and tyrosine kinase inhibitors. The Breast. 2013 Feb;22(1):1-12.
81. Ben-Kasus T, Schechter B, Lavi S, Yarden Y, Sela M. Persistent elimination of ErbB-2/HER2-overexpressing tumors using combinations of monoclonal antibodies: relevance of receptor endocytosis. Proceedings of the National Academy of Sciences of the United States of America. 2009 Mar 3;106(9):3294-9.
82. Logue JS, Morrison DK. Complexity in the signaling network: insights from the use of targeted inhibitors in cancer therapy. Genes & Development. 2012 Apr 1;26(7):641-50.
83. Collins I, Workman P. New approaches to molecular cancer therapeutics. Nature Chemical Biology. 2006 Dec;2(12):689-700.
84. Alexis F. Nano-Polypharmacy to Treat Tumors: Coencapsulation of Drug Combinations Using Nanoparticle Technology. Molecular Therapy. 2014 Jul;22(7):1239-40.
85. Basu S, Chaudhuri P, Sengupta S. Targeting oncogenic signaling pathways by exploiting nanotechnology. Cell Cycle. 2009 Nov 1;8(21):3480-7.
86. Greish K. Enhanced permeability and retention of macromolecular drugs in solid tumors: a royal gate for targeted anticancer nanomedicines. Journal of Drug Targeting. 2007 Aug- Sep;15(7-8):457-64.
87. Hare JI, Lammers T, Ashford MB, Puri S, Storm G, Barry ST. Challenges and strategies in anti-cancer nanomedicine development: An industry perspective. Advanced Drug Delivery Reviews. 2017 Jan 1;108:25-38.
88. Navya PN, Kaphle A, Srinivas SP, Bhargava SK, Rotello VM, Daima HK. Current trends and challenges in cancer management and therapy using designer nanomaterials. Nano Convergence. 2019 Jul 15;6(1):23.
89. Hua S, de Matos MBC, Metselaar JM, Storm G. Current Trends and Challenges in the Clinical Translation of Nanoparticulate Nanomedicines: Pathways for Translational Development and Commercialization. Frontiers in Pharmacology. 2018 Jul 17;9:790.
90. Foroozandeh P, Aziz AA. Insight into cellular uptake and intracellular trafficking of nanoparticles. Nanoscale Research Letters. 2018 Oct 25;13:339.
91. Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annual Review of Biochemistry. 2009 Jul 7;78:857-902.
92. Kumari S, Mg S, Mayor S. Endocytosis unplugged: multiple ways to enter the cell. Cell Research. 2010 Mar;20(3):256-75.
93. Rosenblum D, Joshi N, Tao W, Karp JM, Peer D. Progress and challenges towards targeted delivery of cancer therapeutics. Nature Communications. 2018 Apr 12;9(1):1410.
94. Basu S, Harfouche R, Soni S, Chimote G, Mashelkar RA, Sengupta S. Nanoparticle-mediated targeting of MAPK signaling predisposes tumor to chemotherapy. Proceedings of the National Academy of Sciences of the United States of America. 2009 May 12;106(19):7957-61.
95. Harfouche R, Basu S, Soni S, Hentschel DM, Mashelkar RA, Sengupta S. Nanoparticle-mediated targeting of phosphatidylinositol-3-kinase signaling inhibits angiogenesis. Angiogenesis. 2009 Aug 14;12:325-38.
96. Pi J, Jiang J, Cai H, Yang F, Jin H, Yang P, et al. GE11 peptide conjugated selenium nanoparticles for EGFR targeted oridonin delivery to achieve enhanced anticancer efficacy by inhibiting EGFR-mediated PI3K/AKT and Ras/Raf/MEK/ERK pathways. Drug Delivery. 2017 Nov;24(1):1549-64.
97. Chen K, Zhang Y, Qian L, Wang P. Emerging strategies to target RAS signaling in human cancer therapy. Journal of Hematology & Oncology. 2021 Jul 23;14(1):116.
98. Xue W, Dahlman JE, Tammela T, Khan OF, Sood S, Dave A, et al. Small RNA combination therapy for lung cancer. Proceedings of the National Academy of Sciences of the United States of America. 2014 Aug 26;111(34):E3553-61.
99. Pecot CV, Wu SY, Bellister S, Filant J, Rupaimoole R, Hisamatsu T, et al. Therapeutic Silencing of KRAS Using Systemically Delivered siRNAs. Molecular Cancer Therapeutics. 2014;13(12): 2876-85.
100. Lakshmikuttyamma A, Sun Y, Lu B, Undieh AS, Shoyele SA. Stable and efficient transfection of siRNA for mutated KRAS silencing using novel hybrid nanoparticles. Molecular Pharmaceutics. 2014 Dec 1;11(12):4415-24.
101. Kamerkar S, Lebleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017 Jun 22;546(7659):498-503.
102. Strand MS, Krasnick BA, Pan H, Zhang X, Bi Y, Brooks C, et al. Precision delivery of RAS-inhibiting siRNA to KRAS driven cancer via peptide-based nanoparticles. Oncotarget, 2019 Jul 30;10(46):4761-75.
103. Huang JL, Jiang G, Song QX, Gu X, Hu M, Wang XL, et al. Lipoprotein-biomimetic nanostructure enables efficient targeting delivery of siRNA to Ras-activated glioblastoma cells via macropinocytosis. Nature Communications. 2017 May 10;8:15144.
104. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010 Jan 19;17(1):98- 110.
105. Sheng Z, Evans SK, Green MR. An activating transcription factor 5-mediated survival pathway as a target for cancer therapy? Oncotarget. 2010 Oct;1(6):457-60.
106. Yoon MS, Nanotechnology-Based Targeting of mTOR Signaling in Cancer. International Journal of Nanomedicine. 2020 Aug 6;15:5767-81.
107. Lunova M, Smolková B, Lynnyk A, Uzhytchak M, Jirsa M, Kubinová S, et al. Targeting the mTOR Signaling Pathway Utilizing Nanoparticles: A Critical Overview. Cancers (Basel). 2019 Jan 11;11(1):82.
108. Parveen S, Misra R, Sahoo SK. Nanoparticles: a boon to drug delivery, therapeutics, diagnostics and imaging. Nanomedicine. 2012 Feb;8(2):147-66.
109. Nel AE, Mädler L, Velegol D, Xia T, Hoek EM, Somasundaran P, et al. Understanding biophysicochemical interactions at the nano-bio interface. Nature Materials. 2009 Jul;8(7):543-57.
110. Duan J, Yu Y, Yu Y, Li Y, Wang J, Geng W, et al. Silica nanoparticles induce autophagy and endothelial dysfunction via the PI3K/Akt/mTOR signaling pathway. International Journal of Nanomedicine. 2014 Nov 5;9:5131-41.
111. Chiu HW, Xia T, Lee YH, Chen CW, Tsai JC, Wang YJ. Cationic polystyrene nanospheres induce autophagic cell death through the induction of endoplasmic reticulum stress. Nanoscale. 2015 Jan 14;7(2):736-46.
112. Loos C, Syrovets T, Musyanovych A, Mailänder V, Landfester K, Simmet T. Amino-functionalized nanoparticles as inhibitors of mTOR and inducers of cell cycle arrest in leukemia cells. Biomaterials. 2014 Feb;35(6):1944-53.
113. Hulea L, Markovic Z, Topisirovic I, Simmet T, Trajkovic V. Biomedical Potential of mTOR Modulation by Nanoparticles. Trends in Biotechnology. 2016 May;34(5):349-53.
114. Cai J, Fang L, Huang Y, Li R, Xu X, Hu Z, et al. Simultaneous overactivation of Wnt/ß-catenin and TGFß signalling by miR-128- 3p confers chemoresistance-associated metastasis in NSCLC, Nature Communications. 2017;8:15870.
115. Li J, Yu B, Deng P, Cheng Y, Yu Y, Kevork K, et al. KDM3 epigenetically controls tumorigenic potentials of human colorectal cancer stem cells through Wnt/ß-catenin signalling, Nature Commun. 2017 Apr 25;8:15146.
116. Wan J, Hou X, Zhou Z, Geng J, Tian J, Bai X, et al. WT1 ameliorates podocyte injury via repression of EZH2/ß-catenin pathway in diabetic nephropathy, Free Radical Biology & Medicine. 2017 Jul;108:280-99.
117. Singh S, Mishra A, Mohanbhai SJ, Tiwari V, Chaturvedi RK, Khurana S, et al. Axin-2 knockdown promote mitochondrial biogenesis and dopaminergic neurogenesis by regulating Wnt/ß- catenin signaling in rat model of Parkinson’s disease. Free Radical Biology & Medicine. 2018 Dec;129:73-87.
118. Schneider JA, Craven TW, Kasper AC, Yun C, Haugbro M, Briggs EM, et al. Design of Peptoid-peptide Macrocycles to Inhibit the ß-catenin TCF Interaction in Prostate Cancer. Nature Communications. 2018;9(1):4396.
119. Mariotti L, Pollock K, Guettler S. Regulation of Wnt/ß- catenin signalling by tankyrase-dependent poly(ADP-ribosyl) ation and scaffolding, British Journal of Pharmacology. 2017 Dec;174(24):4611-36.
120. Lee SH, Shin SM, Zhong P, Kim HT, Kim DI, Kim JM, et al. Reciprocal control of excitatory synapse numbers by Wnt and Wnt inhibitor PRR7 secreted on exosomes. Nature Communications. 2018;9(1):3434.
121. Sassi Y, Avramopoulos P, Ramanujam D, Grüter L, Werfel S, Giosele S, et al. Cardiac myocyte miR-29 promotes pathological remodeling of the heart by activating Wnt signaling. Nature Communications. 2017;8(1):1614.
122. Lv C, Li F, Li X, Tian Y, Zhang Y, Sheng X, et al. MiR-31 promotes mammary stem cell expansion and breast tumorigenesis by suppressing Wnt signaling antagonists. Nature Communications. 2017;8(1):1036.
123. Vivero-Escoto JL, Huxford-Phillips RC, Lin W. Silica-based nanoprobes for biomedical imaging and theranostic applications. Chemical Society Reviews. 2012 Apr 7;41(7):2673-85.
124. Malvindi MA, Brunetti V, Vecchio G, Galeone A, Cingolani R, Pompa PP. SiO2 nanoparticles biocompatibility and their potential for gene delivery and silencing. Nanoscale. 2012 Jan 21;4(2):486- 95.
125. Yi H, Wang Z, Li X, Yin M, Wang L, Aldalbahi A, et al. Silica Nanoparticles Target a Wnt Signal Transducer for Degradation and Impair Embryonic Development in Zebrafish. Theranostics. 2016 Jul 18;6(11):1810-20.
126. Valcourt DM, Dang MN, Wang J, Day ES. Nanoparticles for Manipulation of the Developmental Wnt, Hedgehog, and Notch Signaling Pathways in Cancer. Annals Biomedical Engineering. 2020 Jul;48(7):1864-84.
127. Ghoshal A, Goswami U, Sahoo AK, Chattopadhyay A, Ghosh SS. Targeting Wnt Canonical Signaling by Recombinant sFRP1 Bound Luminescent Au-Nanocluster Embedded Nanoparticles in Cancer Theranostics. ACS Biomaterials Science & Engineering. 2015 Nov 2;1(12):1256-66.
128. Gómez-Muñoz A. Ceramide 1-phosphate/ceramide, a switch between life and death. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2006 Dec 1;1758(12):2049- 56.
129. Veeck J, Niederacher D, An H, Klopocki E, Wiesmann F, Betz B, et al. Aberrant methylation of the Wnt antagonist SFRP1 in breast cancer is associated with unfavourable prognosis. Oncogene. 2006 Jun 8;25(24):3479-88.
130. Liu Q, Zhu H, Tiruthani K, Shen L, Chen F, Gao K, et al. Nanoparticle-Mediated Trapping of Wnt Family Member 5A in Tumor Microenvironments Enhances Immunotherapy for B-Raf Proto-Oncogene-Mutant Melanoma. ACS Nano. 2018 Feb 27;12(2):1250-61.
131. Bhattacharyya J, Ren XR, Mook RA, Wang J, Spasojevic I, Premont RT, et al. Niclosamide-conjugated polypeptide nanoparticles inhibit Wnt signaling and colon cancer growth. Nanoscale. 2017 Aug 31;9(34):12709-17.
132. Motawi TK, El-Maraghy SA, ElMeshad AN, Nady OM, Hammam OA. Cromolyn chitosan nanoparticles as a novel protective approach for colorectal cancer. Chemico-Biological Interactions. 2017 Sep 25;275:1-12.
133. Ganesh S, Koser M, Cyr W, Chopda G, Tao J, Shui X, et al. Direct Pharmacological Inhibition of ß-Catenin by RNA Interference in Tumors of Diverse Origin. Molecular Cancer Therapeutics. 2016 Sep;15(9):2143-54.
134. Ma C, Shi L, Huang Y, Shen L, Peng H, Zhu X, et al. Nanoparticle delivery of Wnt-1 siRNA enhances photodynamic therapy by inhibiting epithelial-mesenchymal transition for oral cancer. Biomaterials Science. 2017 Jan 10;5(3):494-501.
135. Riley RS, Day ES. Frizzled7 Antibody-Functionalized Nanoshells Enable Multivalent Binding for Wnt Signaling Inhibition in Triple Negative Breast Cancer Cells. Small. 2017 Jul;13(26):1-20.
136. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nature Reviews Cancer. 2012 Mar 22;12:278-87.
137. Miller-Kleinhenz J, Guo X, Qian W, Zhou H, Bozeman EN, Zhu L, et al. Dual-targeting Wnt and uPA receptors using peptide conjugated ultra-small nanoparticle drug carriers inhibited cancer stem-cell phenotype in chemo-resistant breast cancer. Biomaterials. 2018 Jan;152:47-62.
138. Langer R. Drug delivery and targeting. Nature. 1998 Apr 30;392(6679):5-10.
139. Alexiou C, Arnold W, Klein RJ, Parak FG, Hulin P, Bergemann C, et al. Locoregional cancer treatment with magnetic drug targeting. Cancer Research. 2000 Dec 1;60(23):6641-8.
140. Na HB, Lee JH, An K, Park YII, Park M, Lee IS, et al. Development of a T1 contrast agent for magnetic resonance imaging using MnO nanoparticles. Angewandte Chemie International Edition. 2007;46(28):5397-401.
141. Na HB, Song IC, Hyeon T. Inorganic Nanoparticles for MRI Contrast Agents. Advanced Materials. 2009 Jun 2;21(21):2133-48.
142. Tang M, Russell PJ, Khatri A. Magnetic nanoparticles: prospects in cancer imaging and therapy. Discovery Medicine. 2007;7:68-74.
143. Motoyama J, Yamashita N, Morino T, Tanaka M, Kobayashi T, Honda H. Hyperthermic treatment of DMBA-induced rat mammary cancer using magnetic nanoparticles. BioMagnetic Research and Technology. 2008 Feb 25;6:2.
144. Ciofani G, Riggio C, Raffa V, Menciassi A, Cuschieri A. A bimodal approach against cancer: magnetic alginate nanoparticles for combined chemotherapy and hyperthermia. Medical Hypotheses. 2009;73:80-2..
145. Cedervall T, Lynch I., Lindman S, Berggård T, Thulin E, Nilsson H, et al., Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proceedings og the National Academy of Sciences of the United States of America. 2007 Feb 13;104(7):2050-5.
146. Lynch I, Dawson KA. Protein-nanoparticle interactions. Nanotoday. 2008 Feb-Apr;3(1-2):40-7.
147. Ang JC, Lin JM, Yaron PN, White JW. Protein trapping of silicananoparticles. Soft Matter. 2010;6(2):383-90.
148. Kopp M, Kollenda S, Epple M. Nanoparticle-Protein Interactions: Therapeutic Approaches and Supramolecular Chemistry. Accounts of Chemical Research. 2017 Jun 20;50(6):1383- 90.
149. Park SJ. Protein-Nanoparticle Interaction: Corona Formation and Conformational Changes in Proteins on Nanoparticles. Int J Nanomedicine. 2020 Aug 6;15:5783-802.
150. Huang R, Carney RP, Stellacci F, Lau BLT. Protein-nanoparticle interactions: the effects of surface compositional and structural heterogeneity are scale dependent. Nanoscale. 2013 May 23;5(15):6928-35.
151. Ghosh G, Panicker L, Ningthoujam RS, Barick KC, Tewari R. Counterion induced irreversible denaturation of hen egg white lysozyme upon electrostatic interaction with iron oxide nanoparticles: A predicted model. Colloids and Surfaces B:Biointerfaces. 2013 Mar 1, 103, 267-74.
152. Ghosh G, Panicker L, Barick KC. Protein nanoparticle electrostatic interaction: size dependent counterions induced conformational change of hen egg white lysozyme. Colloids and Surfaces B:Biointerfaces. 2014 Jun 1;118:1-6.
153. Dyawanapelly S, Mehrotra P, Ghosh G, Jagtap DD, Dandekar P, Jain R. How the surface functionalized nanoparticles affect conformation and activity of proteins: exploring through proteinnanoparticles interaction. Bioorganic Chemistry. 2019 Feb;82:17-25.
154. Ghosh G, Panicker L. Protein-nanoparticle interactions and a new insight. Soft Matter 2021 Mar 31, 17(14), 3855-75.
155. Ghosh G. Counterion effects in protein nanoparticle electrostatic binding: A theoretical study. Colloids and Surfaces B: Biointerfaces. 2015 Apr 1;128:23-7.
156. Ghosh G, Gaikwad PS, Panicker L, Nath BB, Mukhopadhyaya R. Unfolding and inactivation of proteins by counterions in proteinnanoparticles interaction. Colloids and Surfaces B: Biointerfaces. 2016 Sep 1;145:194-200.
157. Monopoli MP, Åberg C, Salvati A, Dawson KA. Biomolecular coronas provide the biological identity of nanosized materials. Nature Nanotechnology. 2012 Dec;7(12):779-86.
158. Salvati A, Pitek A, Monopoli M, Prapainop K, Bombelli FB, Hristov DR, et al. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nature Nanotechnology. 2013;8:137-43.
159. Walkey CD, Chan WCW. Understanding and controlling the interaction of nanomaterials with proteins in a physiological environment. Chemical Society Reviews. 2012 Nov 15;41(7):2780-99.
160. Otsuka H, Nagasaki Y, Kataoka K. PEGylated nanoparticles for biological and pharmaceutical applications. Advanced Drug Delivery Reviews. 2003 Feb 24;55(3):403-19.
161. Kim HR, Andrieux K, Delomenie C, Chacun H, Appel M, Desmaële D, et al. Analysis of plasma protein adsorption onto PEGylated nanoparticles by complementary methods: 2-DE, CE and protein lab-on-chip system. Electrophoresis. 2007 Jul;28(13):2252-61.
162. Hamad I, Al-Hanbali O, Hunter AC, Rutt KJ, Andresen TL, Moghimi SM. Distinct polymer architecture mediates switching of complement activation pathways at the nanosphere-serum interface: implications for stealth nanoparticle engineering. ACS Nano. 2010 Nov 23;4(11):6629-38.
163. Schleh C, Semmler-Behnke M, Lipka J, Wenk A, Hirn S, Schäffler M, et al., Size and surface charge of gold nanoparticles determine absorption across intestinal barriers and accumulation in secondary target organs after oral administration. Nanotoxicology. 2012 Feb;6(1):36-46.
164. Choi HS, Ashitate Y, Lee JH, Kim SH, Matsui A, Insin N, et al. Rapid translocation of nanoparticles from the lung airspaces to the body. Nature Biotechnology. 2010 Dec;28(12):1300-3.
165. Oberdörster G, Elder A, Rinderknecht A. Nanoparticles and the brain: cause for concern? Journal of Nanoscience and Nanotechnology. 2009 Aug;9(8):4996-5007.
166. Deng ZJ, Liang M, Monteiro M, Toth I, Minchin RF. Nanoparticle-induced unfolding of fibrinogen promotes Mac-1 receptor activation and inflammation. Nature Nanotechnology. 2011 Dec 19;6:39-44.
167. Marano F, Hussain S, Rodrigues-Lima F, Baeza-Squiban A, Boland S. Nanoparticles: molecular targets and cell signalling. Archives of Toxicology. 2011 Jul;85(7):733-41.
168. Wang J, Jensen UB, Jensen GV, Shipovskov S, Balakrishnan VS, Otzen D, et al. Soft interactions at nanoparticles alter protein function and conformation in a size dependent manner. Nano Letters. 2011 Nov 9;11(11):4985-91.
169. Gagner JE, Lopez MD, Dordick JS, Siegel RW. Effect of gold nanoparticle morphology on adsorbed protein structure and function. Biomaterials. 2011 Oct;32(29):7241-52.
170. Roach P, Farrar D, Perry CC. Surface Tailoring for Controlled Protein Adsorption: Effect of Topography at the Nanometer Scale and Chemistry. J. Am. Chem. Soc. 2006 Mar 7;128(12):3939-45.
171. Mandal HS, Kraatz HB. Effect of the surface curvature on the secondary structure of peptides adsorbed on nanoparticles. Journal of the American Chemical Society. 2007 May 23;129(20):6356-7.
172. Brown DM, Dickson C, Duncan P, Al-Attili F, Stone V. Interaction between nanoparticles and cytokine proteins: impact on protein and particle functionality. Nanotechnology. 2010 Apr 30;21(21):215104.
173. Lacerda SHDP, Park JJ, Meuse C, Pristinski D, Becker ML, Karim A, et al. Interaction of gold nanoparticles with common human blood proteins. ACS Nano. 2010 Dec 18;4(1):365-79.
174. Hussain S, Garantziotis S, Rodrigues-Lima F, Dupret JM, Baeza-Squiban A, Boland S. Intracellular signal modulation by nanomaterials. Advances in Experimental Medicine and Biology. 2014;811:111-34.
175. Rosso F, Marino G, Grimaldi A, Cafiero G, Chiellini E, Chiellini F, et al. Vitronectin absorbed on nanoparticles mediate cell viability/proliferation and uptake by 3T3 Swiss albino mouse fibroblasts: in vitro study. BioMed. Res. Int., 2013;2013:539348.
176. Ghosh G, Mukherjee A, Bhilwade HN, Gupta A, Korde A, Mukhopadhyaya R. Selective Cytotoxicity of Counterion- Conjugated Charged Iron Oxide Nanoparticles: A Study with Lymphoblastoid Raji Cells. J. Adv. Nanomaterials. 2018 Dec;3(4):45-56.
177. Oh E, Delehanty JB, Sapsford KE, Susumu K, Goswami R, Blanco-Canosa JB, et al. Cellular uptake and fate of PEGylated gold nanoparticles is dependent on both cell-penetration peptides and particle size. ACS Nano. 2011 Aug 23;5(8): 6434-48.
178. Wang P, Wang X, Wang L, Hou X, Liu W, Chen C. Interaction of gold nanoparticles with proteins and cells. Sci Technol Adv Mater. 2015 Jun 18;16(3):034610.
179. Zhao F, Zhao Y, Liu Y, Chang X, Chen C, Zhao Y. Cellular uptake, intracellular trafficking, and cytotoxicity of nanomaterials. Small. 2011 May 23;7(10):1322-37.
180. Lunova M, Prokhorov A, Jirsa M, Hof M, Olzynska A, Jurkiewicz P, et al. Nanoparticle core stability and surface functionalization drive the mTOR signaling pathway in hepatocellular cell lines. Scientific Reports. 2017 Nov 22;7:16049.
181. Khan MI, Mohammad A, Patil G, Naqvi SA, Chauhan LK, Ahmad I. Induction of ROS, mitochondrial damage and autophagy in lung epithelial cancer cells by iron oxide nanoparticles. Biomaterials. 2012 Feb;33(5):1477-88.
182. Liu HL, Zhang YL, Yang N, Zhang YX, Liu XQ, Li CG, et al. A functionalized single-walled carbon nanotube-induced autophagic cell death in human lung cells through Akt-TSC2- mTOR signaling. Cell Death & Disease. 2011;2:e159.
183. Kang B, Okwieka P, Schöttler S, Winzen S, Langhanki J, Mohr K, et al. Carbohydrate-Based Nanocarriers Exhibiting Specific Cell Targeting with Minimum Influence from the Protein Corona. Angewandte Chemie International Edition. 2015 Jun 15;54(25):7436-40.
184. Tenzer S, Docter D, Kuharev J, Musyanovych A, Fetz V, Hecht R, et al., Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nature Nanotechnology. 2013 Sep 22;8:772-81.
185. Mirshafiee V, Kim R, Park S, Mahmoudi M, Kraft ML. Impact of protein pre-coating on the protein corona composition and nanoparticle cellular uptake. Biomaterials. 2016 Jan;75:295-304.
186. Corbo C, Molinaro R, Taraballi F, Furman NET, Hartman KA, Shermanet MB, et al. Unveiling the in Vivo Protein Corona of Circulating Leukocyte-like Carriers. ACS Nano. 2017 Mar 28;11(3):3262-73.
187. Schottler S, Becker G, Winzen S, Steinbach T, Mohr K, Landfester K,et al. Protein adsorption is required for stealth effect of poly(ethylene glycol)- and poly(phosphoester)-coated nanocarriers. Nat. Nanotechnol. 2016 Feb 15; 11, 372-7.
188. Takeuchi T, Kitayama Y, Sasao R, Yamada T, Toh K, Matsumoto Y, et al. Molecularly Imprinted Nanogels Acquire Stealth In Situ by Cloaking Themselves with Native Dysopsonic Proteins. Angewandte Chemie International Edition. 2017 Apr 28;129:7194-8.
189. Ke PC, Lin S, Parak WJ, Davis TP, Caruso F. A Decade of the Protein Corona. ACS Nano. 2017 Dec 5;11(12):11773-6.
190. Kah JCY, Chen J, Zubieta A, Hamad-Schifferli K. Exploiting the protein corona around gold nanorods for loading and triggered release. ACS Nano. 2012 Aug 28;6(8):6730-40.
191. Oh JY, Kim HS, Palanikumar L, Go EM, Jana B, Park SA, et al. Cloaking nanoparticles with protein corona shield for targeted drug delivery. Nature Communications. 2018 Oct 31;9:4548.
192. Liu Y, Crawford BM, Vo-Dinh T. Gold nanoparticles-mediated photothermal therapy and immunotherapy. Immunotherapy. 2018 Sep;10(13):1175-88.
193. Chen Y, Gao Y, Chen Y, Liu L, Mo A, Peng Q. Nanomaterialsbased photothermal therapy and its potentials in antibacterial treatment. J. Control Release. 2020 Dec 10;328:251-62.
194. Bardhan R, Lal S, Joshi A, Halas NJ. Theranostic Nanoshells: From Probe Design to Imaging and Treatment of Cancer. Acc. Chem. Res. 2011 May 25;44(10):936-46.
195. Hu X, Zhang Y, Ding T, Liu J, Zhao H. Multifunctional Gold Nanoparticles: A Novel Nanomaterial for Various Medical Applications and Biological Activities. Front. Bioeng. Biotechnol. 2020 Aug 13; 8: 990.
196. Sztandera K, Gorzkiewicz M, Klajnert-Maculewicz B. Gold Nanoparticles in Cancer Treatment. Mol. Pharm. 2019 Jan 7;16(1):1-23.
197. Rastinehad AR, Anastos H, Wajswol E, Winoker JS, Sfakianos JP, Doppalapudi SK, Carrick MR, Knauer CJ, Taouli B, Lewis SC, Tewari AK, Schwartz JA, Canfield SE, George AK, West JL, Halas NJ. Proc. Natl. Acad. Sci. USA. 2019 Sep 10;116(37):18590-18596.
198. Fernandes N, Rodrigues CF, Moreira AF, Correia IJ. Overview of the application of inorganic nanomaterials in cancer photothermal therapy. Biomater Sci. 2020 Apr 15;8(11):2990-3020.
199. Estelrich J, Busquets MA. Iron Oxide Nanoparticles in Photothermal Therapy. Molecules. 2018 Jun 28;23(7):1567.
200. Sharma S, Chakraborty N, Jha D, Gautam HK, Roy I. Robust dual modality antibacterial action using silver-Prussian blue nanoscale coordination polymer. Mater. Sci. Eng. C Mater. Biol. Appl. 2020 Aug;113:110982.
201. Chakraborty N, Jha D, Gautam HK, Roy I. Peroxidase-like behavior and photothermal effect of chitosan coated Prussianblue nanoparticles: dual-modality antibacterial action with enhanced bioaffinity. Mater. Adv. 2020 May 27;1:774-84.
202. Yang M, Li J, Gu P, Fan X. The application of nanoparticles in cancer immunotherapy: Targeting tumor microenvironment. Bioact. Mater. 2020 Dec 26;6(7):1973-87.
203. Hou X, Tao Y, Pang Y, Li X, Jiang G, Liu Y. Nanoparticle-based photothermal and photodynamic immunotherapy for tumor treatment. Int. J. Cancer. 2018 Dec 15;143(12):3050-60.
204. Rahban D, Doostan M, Salimi A. Cancer Therapy; Prospects for Application of Nanoparticles for Magnetic-Based Hyperthermia. Cancer Invest. 2020 Sep 14;38(8-9):507-21.
205. Das P, Colombo M, Prosperi D. Recent advances in magnetic fluid hyperthermia for cancer therapy. Colloids Surf. B:Biointerfaces. 2019 Feb 1;174:42-55.
206. Tomitaka A, Takemura Y. Magnetic Relaxation of Intracellular Magnetic Nanoparticles for Hyperthermia. Crit. Rev. Biomed. Eng. 2019;47(6), 489-94.
207. Zhu L, Zhou Z, Mao H, Yang L. Magnetic nanoparticles for precision oncology: theranostic magnetic iron oxide nanoparticles for image-guided and targeted cancer therapy. Nanomedicine (Lond). 2017 Jan;12(1):73-87.
208. Lyer S, Singh R, Tietze R, Alexiou C. Magnetic nanoparticles for magnetic drug targeting. Biomed. Tech. (Berl). 2015 Oct 1;60(5):465-75.
209. Maier-Hauff K, Ulrich F, Nestler D, Niehoff H, Wust P, Thiesen B, Orawa H, Budach V, Jordan A. Efficacy and safety of intratumoral thermotherapy using magnetic iron-oxide nanoparticles combined with external beam radiotherapy on patients with recurrent glioblastoma multiforme. J. Neurooncol. 2011 Jun;103(2):317-24
210. Gandhi S, Issar S, Mahapatro AK, Roy I. Cobalt ferrite nanoparticles for bimodal hyperthermia and their mechanistic interactions with lysozyme. J. Mol. Liq. 2020 Jul 15;310:113194.
211. Moy AJ, Tunnell JW. Combinatorial immunotherapy and nanoparticle mediated hyperthermia. Adv. Drug Deliv. Rev. 2017 May 15;114:175-83.