Abstract
The desmosomal cadherin Desmoglein-3 (Dsg3) is a core adhesion component in desmosome junctions that occur with high frequency in the stratified squamous epithelial membrane lining the skin and mucous membrane. Dsg3 is identified as a major target of the circulating autoantibodies in Pemphigus Vulgaris (PV), an autoimmune blistering skin disease, and many signaling pathways have been demonstrated to be activated by PV-IgG targeting Dsg3, highlighting its role as a surface regulator in cell signaling. A recent study has revealed an unprecedented role of Dsg3 in the suppression of p53 and shows dysfunction of this pathway in PV. Furthermore, reciprocal crosstalk between p53 and yes-associated protein (YAP) downstream of Dsg3 has been observed in keratinocytes in which increased YAP expression causes suppression of p53 or vice versa. Both p53 and YAP are the crucial nuclear transcription factors involved in regulating cell fate decision, adaptation and tissue integrity in response to environmental and biological cues and are mutually exclusive in human cancer. In this review, we discuss Dsg3 signaling role in keratinocyte response to stress signals, with the highlight on our recent findings of the Dsg3/p53 pathway in the control of cell proliferation and tissue homeostasis, including the DNA integrity, beyond its function in cell-cell adhesion.
Keywords
Desmoglein-3, p53, YAP, Cell signaling, DNA damage, Pemphigus
Introduction
Desmoglein-3 (Dsg3) belongs to a subfamily of the desmosomal cadherins and is an essential component of the junctional protein complex known as the desmosome that mediates calcium-dependent cell-cell adhesion in vertebrate epithelial cells [1]. Desmosomes occur in abundance in tissues, such as the skin and mucous membrane that are subjected to extensive mechanical stress. In addition to its role in cell-cell adhesion, Dsg3 also functions as a surface regulator for various intracellular signaling pathways in epithelial cells [1-7]. Many of these findings are achieved from the studies of the pathogenesis of Pemphigus Vulgaris (PV), an autoimmune bullous disease in which Dsg3 serves as a major autoantigen and is targeted by circulating autoantibodies that cause disruption of desmosomes, resulting in blistering affecting both the skin and mucous membrane [2,3,6]. This mini-review will focus on our recent findings suggesting an unprecedented signaling role of Dsg3 in regulating two fundamental pathways that control cell proliferation and cell fate decision [8,9]. The involvement of this pathway in the pathogenesis of PV is also discussed briefly in this review.
Dsg3 Exerts Function as a Cell Signaling Regulator
Several studies have implicated a key role for Dsg3 in mediating outside-in signaling (e.g. Src, p38MAPK, PKC, ERK, EGFR, plakoglobin, c-Myc, and Rho GTPases, etc.) involved in desmosome remodeling, cell proliferation, differentiation, migration or apoptosis, and thus validating that Dsg3 acts as a signaling molecule that has a major impact on tissue integrity and homeostasis [1]. Early studies have shown activation of phospholipase C signaling pathway following the binding of PV-IgG to Dsg3 that leads to disruption of cell cohesion and blister formation [10]. Dysregulation of Dsg3 in the suprabasal layers of the skin in the transgenic mouse resulted in increased keratinocyte proliferation and altered terminal differentiation suggesting that this basal cell isoform of Dsg is involved in the regulation of cell proliferation [11]. Consistent with this finding, another study utilizing Dsg3 depletion in HaCaT keratinocytes demonstrated the suppression of cell proliferation and colony growth [12]. Such an effect of altered Dsg3 expression on cell proliferation may indicate a link between Dsg3 loss and cell cycle control [13]. Owing to the widespread expression of Dsg3 at the plasma membrane, it is thought that the non-junctional pool of Dsg3 may be responsible for cell signaling and is the target of PV-IgG [14,15]. Tsang et al. have demonstrated that non-junctional Dsg3 acts as an upstream regulator of Src via interaction with classical E-cadherin and helps to regulate the formation of adherens junctions [15-17]. Overexpression of Dsg3 in epidermoid carcinoma A431 cell line elicits activation of Ezrin and AP-1 that are associated with tumor cell migration and invasion [18]. Dsg3 is also found to act as an upstream regulator of small Rho GTPases and overexpression of Dsg3 results in increased activity of Rac1 and Cdc42 in epithelial cells [17]. In line with this finding, pemphigus IgG binding to Dsg3, as well as Desmoglein-1 (Dsg1), interferes with RhoA activity and signaling that is coupled with a range of PV-IgG triggered effects, including keratin retraction and release of Dsg3 from the cytoskeleton-associated protein pool [19,20]. Activation of all these signaling pathways can have an influence on E-cadherin trafficking and the modulation of E-cadherin mediated junction stability [21]. Besides, numerous intracellular signalling events are found to be activated by IgG binding to Dsg3 [2,3,5,6], among which is the key pathway p38MAPK. Enhanced phosphorylation of p38MAPK and its downstream target HSP27 was detected in keratinocytes treated with PV-IgG and the pathogenic anti-Dsg3 antibody AK23 [22,23]. Pathways such as PKC, EGFR, Src and c-Myc, among many others, have also been implicated in PV pathological signaling events [3,4,23,24]. Inhibition of these pathways can alleviate blistering in vitro and in vivo [5,25-27]. Together, these data suggest that Dsg3 is an important signaling molecule in human keratinocytes.
Dsg3 Regulates the p53 Pathway
The Dsg3 bearing tissues such as skin and oral mucosa are constantly exposed to a multitude of stresses due to external and internal influences that can potentially lead to genome damage and cellular oxidative stress [28-30]. p53 is a central hub in response to all these stress signals via coordinating many cellular stress responses to maintain genomic integrity and hence is called a ‘cellular gatekeeper’ [31] and the ‘guardian of the genome’ [32]. Several studies have highlighted the connection between altered cellular stress response and human diseases, including diabetes and cancer [33]. Ultraviolet (UV) irradiation can induce the generation of reactive oxidative species (ROS) [34]. Oxidative stress is shown to be involved in numerous skin diseases including psoriasis, lichen planus and pemphigus [35-38]. Moreover, p53 overexpression is reported in skin conditions such as psoriasis [39], discoid lupus erythematosus (DLE) [40], and pemphigus in two studies with a focus on apoptosis [41,42]. Nonetheless, to our best knowledge, no study to date has explored the direct functional association between Dsg3 and the p53 pathway except for our recent report [8].
Taking into account that 1) Dsg3 acts as a cell signal regulator for various pathways, 2) our recent study shows that Dsg3 exerts a function as a sensor to mechanical stress in keratinocytes [9], and 3) the Dsg3 bearing tissues are prone to various chemical and physical stresses daily that could induce the p53 activation [43], we performed a study with a focus on the establishment of the Dsg3/p53 pathway using various in vitro and in vivo study approaches, including Dsg3 knockdown and knockout mouse model and analysis of PV patient specimens as well as keratinocyte cultures treated with PV-IgG and the specific anti-Dsg3 antibody [8]. This study sheds new light on Dsg3’s ability as an anti-stress protein by restricting p53 response to stress signals in keratinocytes. The study demonstrated moderate, but significant elevated p53 expression in cells harboring wild type p53, with Dsg3 depletion mediated by ribonucleic acid interference (RNAi) and this was accompanied by an increased expression of p21Waf1/Cip1 and Bax suggesting activation of the p53 pathway (Figure 1). The correlation of increased p21Waf1/Cip1 with p53 activation in Dsg3 depleted cells was further elaborated by knocking down of p53 that resulted in p21Waf1/Cip1 abrogation. The p53 response to stress involves the coordination of cell cycle arrest and apoptosis [43] and p53 accomplishes these effects by regulating several genes, primarily including p21Waf1/Cip1 and Bax. p21Waf1/Cip1 is upregulated by p53 following DNA damage [44-46] and is thought to be an integral part of the p53-mediated growth-arrest pathway [45]. Besides, Bax is a p53 primary response gene involved in a p53 regulated apoptotic activity by both transcriptiondependent and independent pathways [47,48]. Thus, the combined upregulation of p53 along with p21Waf1/Cip1 and Bax indicates activation of the p53 pathway. These in vitro findings were supported by in vivo study in Dsg3 null mice that showed increased expression of p53/p21Waf1/Cip1/cleaved caspase-3 in Dsg3−/− hair follicles in dorsal skin samples, but not in Dsg3+/− littermates [8]. Notably, we showed that the Dsg3/p53 pathway is Dsg3 specific as neither E-cadherin nor desmoplakin knockdown elicits the same effect. Furthermore, this pathway only applies to wild type p53 since modulation of Dsg3 expression in A431 cell line that bears p53 mutation failed to induce any changes in p53 with no detection of p21Waf1/Cip1. Collectively, these findings are consistent with the hypothesis that Dsg3 plays a role in dampening the p53 response to stress that may be required to control the balance of the p53 pathway in epithelial cells, in particular in stress-baring tissues.
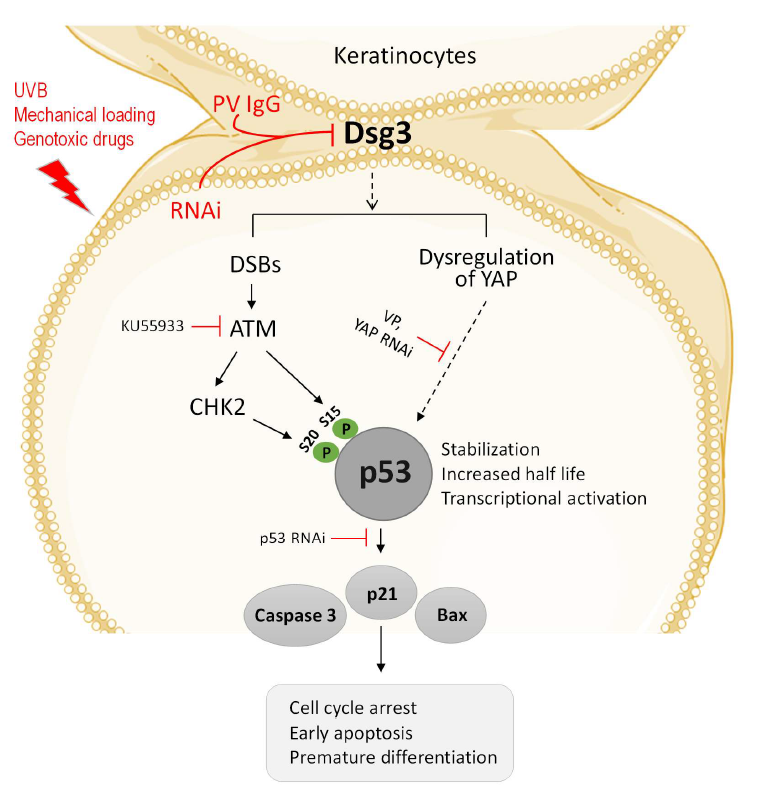
The stabilization of p53 is a common response to cellular stress and is one of the key mechanisms by which the p53 function is regulated. Many tumors that retain wild type p53 show defects in this pathway [49]. The stabilization of p53 also seems to be the case in Dsg3 depleted cells since Dsg3 knockdown resulted in an increase of the half-life of p53 protein turnover by approximately two-fold. We showed that such delayed p53 turnover was accompanied by stabilization of MDM2 that may reflect a negative feedback mechanism to control the p53 expression levels [50]. It is worth noting that the stress response of p53 can be obscured by the nature of its fast turnover as we demonstrated by treating cells with the proteasome inhibitor MG132 that revealed greater differences in the levels of p53 expression between the Dsg3 knockdown and control cells [8]. Consistent with the findings from the loss-of-function study, overexpression of Dsg3 resulted in an inverse effect with marked suppression of p53 at both protein and mRNA levels as well as the p53 transcription activity [8]. Furthermore, this regulatory pathway of Dsg3/p53 was consolidated by experiments with several cellular stress responses, such as UV irradiation, genotoxic drug treatment and cyclic mechanical strain, which provoked further enhancement in the levels of p53 and its downstream targets p21Waf1/Cip1 and Bax in cells with Dsg3 knockdown [8]. Collectively, these results suggest strongly that Dsg3 indeed functions as a sensor by modulating the p53 response to stress signals.
Dsg3 Knockdown Results in DNA Damage
Our recent study also identified the activation of the Ataxia-telangiectasia mutated (ATM) pathway following Dsg3 depletion in the UV setting (Figure 1) [8]. ATM is activated by DNA damage and stress [51] and plays a key role in regulating the DNA damage response. ATM is primarily involved in detecting DNA double-strand breaks (DSBs), leading ultimately to the direct or indirect (via CHK2) activation and stabilization of p53 [52]. DNA DSBs can be detected by the presence of either γ-H2AX or p53-binding protein 1 (53BP1) [53]. Our study identified significant induction of 53BP1 in Dsg3 depleted cells, especially in those with UV exposure, and this was accompanied by upregulation of phosphorylated, as well as total, ATM, CHK2, and p53-S15/20, compared to the respective controls [8]. This data suggests that Dsg3 depletion somehow potentiates UV induced DNA damage with increased DNA DSBs, albeit the underlying mechanism remains obscure. It was thought that this could well be due to the oxidative stress caused by disruption of the intercellular junctions.
Involvement of the Dsg3/p53 Pathway in the Pathogenesis of PV
PV is a rare, severe autoimmune mucocutaneous blistering disease characterized by autoantibodies (PV-IgG) targeting the desmosome cell adhesion complexes in keratinocytes, leading to loss of cell-cell adhesion (acantholysis) [3,5]. Patients with PV may suffer from malnutrition or even multiple life-threatening complications associated with certain therapies, including bacterial and viral infection, fluid and electrolyte imbalances, and cardiac and renal failure [54,55]. The pathogenesis of PV remains not completely clear, especially for the key driver in blister formation. It was thought that multiple factors are involved in blistering, including steric hindrance, several intracellular signaling events (e.g. Src, p38MAPK, PKC. ERK, etc.), Dsg3 degradation, cell shrinkage, apoptosis and also the genetic factors that facilitates the production of autoantibodies against Dsg3 [5,56]. Autoantibodies binding to the adhesion site in the extracellular domain of Dsg3 is considered the initial and critical step in the pathogenesis of PV, leading to the disruption and loss of cell cohesion and ultimately blistering in the skin and oral mucosa [2-5]. However, the direct inhibition of Dsg3 binding is not sufficient to induce the complete loss of cell cohesion [57,58]. Thus, the synergistic action of different pathways/events is considered to contribute to blistering in this disease [3,5]. Many signaling pathways are identified to contribute to the loss of keratinocyte cohesion, such as Src, p38MAPK, EGFR, c-Myc, and Rho GTPases [3,5,15-17,25,58-63]. Moreover, studies also have shown an increased incidence of apoptotic occurrence in acantholysis lesions of PV [41,64]. Elevated expression of pro-apoptotic proteins Bax, FasL, and caspases, as well as downregulation of the anti-apoptotic Bcl-2, have been reported in the literature [65-69]. Together, these studies have indicated that Dsg3 serves as a key receptor to transmit outside-in signaling that results in blister formation in PV. However, the exact role of how Dsg3 mediates intracellular signaling is not fully understood. Moreover, studies have also suggested that the pathology of PV is caused by autoantibodies targeting non-Dsg3 receptors on the cell surface, thereby triggering intracellular signaling and leading to apoptosis and ultimately, blistering [70,71].
To investigate the potential involvement of the Dsg3/ p53 pathway in the pathogenesis of PV, we examined the biopsies from patients with PV. We showed ~50% of PV patients having a markedly elevated expression of p53 as well as cleaved caspase-3, indicating early apoptosis [8]. Importantly, these changes were found not only in cells surrounding the blisters but also in perilesional regions, suggesting that activation of the p53 pathway likely occurs early before the event of pemphigus acantholysis. Since each patient with PV may produce a unique composition of autoantibodies that triggers a particular pattern of signaling events [72], the observed alterations in p53 expression were not surprising, and this could be due to the variations of the disease status and the effect of the treatment received, or additionally because p53 in response to stress is transient and declines over time [73]. Consistent with this notion, a previous study using PV– IgG to treat keratinocytes and skin organ cultures showed a 50% and 40% increase of p53, respectively, compared to controls [41].
The specificity of p53 induction elicited by PV-IgG targeting Dsg3 was verified by additional in vitro studies with PV sera that contain a pool of anti-Dsg3 antibodies (i.e. polyclonal antibodies) and also with the well-characterized specific pathogenic monoclonal antibody AK23 that binds the Dsg3 adhesion site at the N-terminal [74]. The results from both experimental approaches indicated a marked increase of p53 as well as Bax, concomitant with Dsg3 depletion as expected based on several previous studies with PV sera [14,75]. Thus, the observed findings in PV indicate a specific p53 induction associated with PV-IgG induced Dsg3 disturbance as this effect was demonstrated by the treatment of cells with anti-Dsg3 antibody AK23 in a time and dose-dependent manners [8]. Furthermore, it was proved that enhanced p53 is specific since the RNAi mediated p53 knockdown significantly abated the PV sera induced positive p53 signals. These results suggest strongly that activation of the Dsg3/p53 pathway may contribute, at least in part, to PV pathology, with the evidence of early apoptosis that has been shown by others [65,68,76]. This finding may have important implications in clinical diagnosis and also in the development of a novel therapeutic strategy in treating this life-threatening autoimmune disease in the future.
Dsg3 is Involved in the Regulation of YAP
Our recent report also shows that Dsg3 is involved in the regulation of yes-associated protein (YAP) via forming a protein complex with phospho-YAP in HaCaT keratinocyte cell line [9]. Knocking down of Dsg3 has an impact on YAP expression at both the transcript and protein levels and causes the failure of YAP response to cyclic mechanical strain [77]. The same results were observed in N/TERT keratinocytes and showed that Dsg3 forms a protein complex with both YAP and phospho-YAP in this cell line (our unpublished data). Dsg3 depletion in N/TERTs also resulted in a marked reduction of both YAP and phospho- YAP. These results collectively indicate a permissive role for Dsg3 in the regulation of the YAP-Hippo pathway in keratinocytes. The Hippo pathway is another essential pathway in the control of cell differentiation and organ growth [78,79]. A key downstream nuclear effector and transcription coactivator of this pathway is YAP [77,79,80]. The activity of YAP is partially regulated by protein phosphorylation, including serine residue S127, leading to YAP nuclear exclusion and inactivation through the process of degradation or cytoplasmic accumulation via binding to 14-3-3 [79]. However, identification of the upstream regulators of the Hippo pathway remains limited, and especially, there is still a lack of the candidates for transmembrane proteins. Our findings suggest that Dsg3 is a potential transmembrane protein that regulates Hippo, which warrants further investigation.
p53 and YAP Antagonize Each Other Downstream of Dsg3
In an attempt to address the involvement of YAP in the Dsg3/p53 pathway, we performed experiments by knocking down of YAP or treating cells with the YAP inhibitor, and our results indicated both approaches caused increased p53 expression and nuclear accumulation (data not shown) (Figure 1). In contrast, transfection of YAP into the Dsg3 depleted cells partially rescued the phenotype of the p53 induction. Furthermore, the antagonistic regulation between YAP and p53 was demonstrated by p53 Luciferase assay that showed inhibition of p53 transcription activity in cells with YAP transfection, with an inverse effect observed in cells with YAP knockdown, as compared to the respective controls. Hence, it is speculated that YAP may bridge in or have an influence on the Dsg3/p53 pathway in keratinocytes, as shown in a working model that Dsg3 restricts p53 via YAP. This simplified model illustrates a relationship among these three signaling molecules in keratinocyte response to stress signals (Figure 1). Notably, this model places Dsg3 upstream of p53 and YAP and indicates that modulation of Dsg3 could have an impact on both signaling pathways, highlighting Dsg3 as an important component of the cellular stress response network in keratinocytes. Hence, this is another example among many other upstream regulators that elaborates reciprocal crosstalk between YAP and p53 for fine-tuning of cell proliferation and apoptosis [81,82].
The central hub in control of the balance between cellular proliferation and differentiation involves both fundamental pathways, p53 and Hippo-YAP signaling [78,83]. A previous study by our group has shown that modulation of Dsg3 has an impact on cell proliferation and Dsg3 silencing mediated by RNAi impaired cell proliferation and colony expansion in HaCaT keratinocyte line [12]. However, the underlying molecular mechanism was unclear, and it was thought that this could be due to a defect in cell-cell adhesion. Now, it becomes clear based on our recent findings [8,9] that this is caused by a reduction of YAP and suppression of YAP activity following Dsg3 depletion. p53 might also play a role here in Dsg3 siRNA treated HaCaT cells as p53 was reported still to be functional despite its point mutations in this cell line [84]. Consistent with the notion that Dsg3 regulates cell proliferation via suppression of p53, overexpression of Dsg3 suppresses cell differentiation (suggesting cell de-differentiation) and knockdown of Dsg3 results in an increase of various early keratinocyte differentiation markers (suggesting premature cell differentiation) in several cell lines [8,85]. As p53 is also a well-characterized regulator in cell differentiation [86,87], it is not surprising that alteration of p53 can have an impact on the balance of cellular proliferation and differentiation.
Conclusion and Future Perspectives
In summary, our recent studies provide novel evidence that Dsg3 plays a role in regulating p53 response to stress signals in keratinocytes and this pathway likely involves YAP that acts in the suppression of the p53 pathway. Alterations of this pathway may attribute to the pathogenesis of PV where Dsg3 is targeted by autoantibodies resulting in its degradation (loss of function), leading to heightened p53 levels and the activation of the apoptotic machinery. As a consequence, disruption of cell adhesion and cell shrinkage occurs that causes blistering in the Dsg3 baring tissues. The direct evidence of p53 in PV was lacking, and this study fills this gap, suggesting that Dsg3 signaling towards p53 likely reflects cellular stress response in PV. Hence this finding underscores a central role for Dsg3 in pemphigus pathogenesis. The finding also advances our understanding of normal physiological conditions. For instance, the distinct expression patterns of Dsg3 between the skin and mucous membrane may reflect their exposures to different environmental stresses and insults. While the oral mucous membrane is subject to physical and chemical insults daily, the skin is readily prone to UV irradiation with relative less frequency of mechanical stimulation (e.g. trunk skin). Thus this study sheds a light on a potential role for Dsg3 in control of tissue integrity (including the DNA) and homeostasis in these stress-bearing tissues and indicates the pivotal function of Dsg3 as a stress sensor and responder in keratinocytes beyond cell-cell adhesion.
Overall, our study reveals an unprecedented role of Dsg3 in keratinocyte stress response via regulating the p53 and YAP pathways that paves the way for further research, especially on an emerging concept about the junctional proteins such as Dsg3 in control of the integrity of junctional network as well as the cell fate decision. An avenue to further explore is how Dsg3 is able to sense stress. An interesting finding from Kathleen Green’s group/ laboratory has shown suppression of Dsg1, among other early differentiation proteins, in response to UV irradiation [88]; thus downregulation of Dsg1 in response to stress might potentially have influence on the Dsg3/p53 pathway, and further work in this aspect will help to establish this notion. Another possible avenue for future research is to characterize the functional association of Dsg3 and YAP by investigating if the interaction is direct or indirect via other scaffolding protein(s). One of our intriguing observations was that knockdown of Dsg3 resulted in the loss of membrane localization of phospho-YAP in keratinocytes with an effect of the nuclear retention in Dsg3 knockdown cells, especially in HaCaT cell line [9]. Previous studies have identified that the phosphorylation of YAP at S128 caused by Nemo-Like Kinase (NLK) is the key for YAP nuclear localization [89,90]. Thus, it was speculated that Dsg3 might negatively regulate the phosphorylation of YAP-S128 by suppressing NLK. It would, therefore, be interesting to analyze the phosphorylation at YAP-S128 and NLK and this additional information may help to elucidate whether the phosphorylation at S128 is regulated by Dsg3 and is required for YAP nuclear relocation in Dsg3 depleted cells. Finally, in order to pursue the presented findings in PV towards the translational research with a clinical application being beneficial to patients, further studies are required to gain the mechanistic insight on the identified p53 signaling. Thus, insights into the activation of the apoptotic pathway in a spatial-temporal manner in the disease process of PV and also in the correlation between p53 induction and disease activity are necessary.
Abbreviations
53BP1: p53-binding Protein 1; ATM: Ataxiatelangiectasia Mutated; Bcl-2: B-cell lymphoma-2; DLE: Discoid Lupus Erythematosus; DSB: DNA double-strand Breaks; Dsg1: Desmoglein-1, Dsg: Desmoglein-3; ERK: Extracellular signal-regulated Kinase; EGFR: Epidermal Growth Factor Receptor; FasL: Fas Ligand; mRNA: messenger Ribonucleic Acid; NLK: Nemo-like Kinase; PCNA: Proliferating Cell Nuclear Antigen; p38MAPK: p38 Mitogen-activated Protein Kinases; PKC: Protein Kinase C; PV: Pemphigus Vulgaris; RNAi: Ribonucleic Acid Interference; ROS: Reactive Oxygen Species; UV: Ultraviolet; VP: Verteporfin; YAP: Yes-associated Protein
Acknowledgements
We would like to thank all the members of Dr Wan’s group, including Yunying Huang, Jutamas Uttagomol and Usama Sharif Ahmad for their hard work and contributions to this work. We also want to express special thanks to our international collaborators, Yang Cai, Hana Jedlickovah, Martin Röcken, and Christian Hünefeld for their valuable contributions, and colleagues in Blizard institute, including Eric Kenneth Parkinson, Catherine Harwood, Daniele Bergamaschi, Muy Teck The, Angray Kang, Ankit Patel, Gary Warnes, Jan Soetaert and Belen Martin-Martin for helpful discussions, advice and assistance throughout this project’s research. We also are very grateful to Ian R. Hart for assisting the editing of the original research articles. This work was supported by the Institute of Dentistry, Barts and The London School of Medicine and Dentistry and Naresuan University, Thailand.
Declaration of Competing Interest
The authors declare that there is no conflict of interests that could have influenced to this work.
References
2. Hammers CM, Stanley JR. Mechanisms of disease: pemphigus and bullous pemphigoid. Annual Review of Pathology: Mechanisms of Disease. 2016 May 23;11:175- 97.
3. Spindler V, Eming R, Schmidt E, Amagai M, Grando S, Jonkman MF, et al. Mechanisms causing loss of keratinocyte cohesion in pemphigus. Journal of Investigative Dermatology. 2018 Jan 1;138(1):32-7.
4. Kitajima Y. 150th anniversary series: desmosomes and autoimmune disease, perspective of dynamic desmosome remodeling and its impairments in pemphigus. Cell Communication & Adhesion. 2014 Dec 1;21(6):269-80.
5. Ahmed AR, Carrozzo M, Caux F, Cirillo N, Dmochowski M, Alonso AE, et al. Monopathogenic vs multipathogenic explanations of pemphigus pathophysiology. Experimental Dermatology. 2016 Nov;25(11):839-46.
6. Takahashi H, Iriki H, Mukai M, Kamata A, Nomura H, Yamagami J, et al. Autoimmunity and immunological tolerance in autoimmune bullous diseases. International Immunology. 2019 Jul;31(7):431-7.
7. Brown L, Wan H. Desmoglein 3: a help or a hindrance in cancer progression?. Cancers. 2015 Mar;7(1):266-86.
8. Rehman A, Cai Y, Hunefeld C, Jedlickova H, Huang Y, Teck TM, et al. The desmosomal cadherin desmoglein-3 acts as a keratinocyte anti-stress protein via suppression of p53. Cell Death and Disease. 2019 Oct 3;10(10):1-4.
9. Uttagomol J, Ahmad US, Rehman A, Huang Y, Laly AC, Kang A, et al. Evidence for the Desmosomal Cadherin Desmoglein-3 in Regulating YAP and Phospho- YAP in Keratinocyte Responses to Mechanical Forces. International Journal of Molecular Sciences. 2019 Jan;20(24):6221.
10. Kitajima Y. New insights into desmosome regulation and pemphigus blistering as a desmosome-remodeling disease. The Kaohsiung Journal of Medical Sciences. 2013 Jan 1;29(1):1-13.
11. Merritt AJ, Berika MY, Zhai W, Kirk SE, Ji B, Hardman MJ, et al. Suprabasal desmoglein 3 expression in the epidermis of transgenic mice results in hyperproliferation and abnormal differentiation. Molecular and Cellular Biology. 2002 Aug 15;22(16):5846-58.
12. Mannan T, Jing S, Foroushania SH, Fortune F, Wan H. RNAi-mediated inhibition of the desmosomal cadherin (desmoglein 3) impairs epithelial cell proliferation. Cell Proliferation. 2011 Aug;44(4):301-10.
13. Chen YJ, Lee LY, Chao YK, Chang JT, Lu YC, Li HF, et al. DSG3 facilitates cancer cell growth and invasion through the DSG3-plakoglobin-TCF/LEF-Myc/cyclin D1/MMP signaling pathway. PloS One. 2013 May 30;8(5):e64088.
14. Aoyama Y, Kitajima Y. Pemphigus vulgaris-IgG causes a rapid depletion of desmoglein 3 (Dsg3) from the Triton X-100 soluble pools, leading to the formation of Dsg3-depleted desmosomes in a human squamous carcinoma cell line, DJM-1 cells. Journal of Investigative Dermatology. 1999 Jan 1;112(1):67-71.
15. Tsang SM, Brown L, Lin K, Liu L, Piper K, O’Toole EA, et al. Non-junctional human desmoglein 3 acts as an upstream regulator of Src in E-cadherin adhesion, a pathway possibly involved in the pathogenesis of pemphigus vulgaris. The Journal of Pathology. 2012 May;227(1):81-93.
16. Tsang SM, Liu L, Teh MT, Wheeler A, Grose R, Hart IR, et al. Desmoglein 3, via an interaction with E-cadherin, is associated with activation of Src. PLoS One. 2010 Dec 3;5(12):e14211.
17. Tsang SM, Brown L, Gadmor H, Gammon L, Fortune F, Wheeler A, et al. Desmoglein 3 acting as an upstream regulator of Rho GTPases, Rac-1/Cdc42 in the regulation of actin organisation and dynamics. Experimental Cell Research. 2012 Nov 1;318(18):2269-83.
18. Brown L, Waseem A, Cruz IN, Szary J, Gunic E, Mannan T, et al. Desmoglein 3 promotes cancer cell migration and invasion by regulating activator protein 1 and protein kinase C-dependent-Ezrin activation. Oncogene. 2014 May;33(18):2363-74.
19. Waschke J, Spindler V, Bruggeman P, Zillikens D, Schmidt G, Drenckhahn D. Inhibition of Rho A activity causes pemphigus skin blistering. The Journal of Cell Biology. 2006 Dec 4;175(5):721-7.
20. Spindler V, Waschke J. Role of Rho GTPases in desmosomal adhesion and pemphigus pathogenesis. Annals of Anatomy-Anatomischer Anzeiger. 2011 May 1;193(3):177-80.
21. Moftah H, Dias K, Apu EH, Liu L, Uttagomol J, Bergmeier L, Kermorgant S, Wan H. Desmoglein 3 regulates membrane trafficking of cadherins, an implication in cell-cell adhesion. Cell Adhesion & Migration. 2017 May 4;11(3):211-32.
22. Kawasaki Y, Aoyama Y, Tsunoda K, Amagai M, Kitajima Y. Pathogenic monoclonal antibody against desmoglein 3 augments desmoglein 3 and p38 MAPK phosphorylation in human squamous carcinoma cell line. Autoimmunity. 2006 Jan 1;39(7):587-90.
23. Saito M, Stahley SN, Caughman CY, Mao X, Tucker DK, Payne AS, et al. Signaling dependent and independent mechanisms in pemphigus vulgaris blister formation. PloS One. 2012 Dec 3;7(12):e50696.
24. Kugelmann D, Rötzer V, Walter E, Egu DT, Fuchs MT, Vielmuth F, et al. Role of Src and cortactin in pemphigus skin blistering. Frontiers in Immunology. 2019 Apr 4;10:626.
25. Walter E, Vielmuth F, Rotkopf L, Sárdy M, Horváth ON, Goebeler M, et al. Different signaling patterns contribute to loss of keratinocyte cohesion dependent on autoantibody profile in pemphigus. Scientific Reports. 2017 Jun 15;7(1):3579.
26. Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA, Rubenstein DS. p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proceedings of the National Academy of Sciences. 2006 Aug 22;103(34):12855-60.
27. Berkowitz P, Hu P, Liu Z, Diaz LA, Enghild JJ, Chua MP, et al. Desmosome signaling. Inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. Journal of Biological Chemistry. 2005 Jun 24;280(25):23778-84.
28. Ashcroft M, Taya Y, Vousden KH. Stress signals utilize multiple pathways to stabilize p53. Molecular and Cellular Biology. 2000 May 1;20(9):3224-33.
29. Haigis KM, Sweet-Cordero A. New insights into oncogenic stress. Nature Genetics. 2011 Mar;43(3):177-8.
30. Sinha RP, Häder DP. UV-induced DNA damage and repair: a review. Photochemical & Photobiological Sciences. 2002;1(4):225-36.
31. Levine AJ. p53, the cellular gatekeeper for growth and division. cell. 1997 Feb 7;88(3):323-31.
32. Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15-6.
33. Fulda S, Gorman AM, Hori O, Samali A. Cellular stress responses: cell survival and cell death. International journal of cell biology. 2010 Oct;2010:214074.
34. de Jager TL, Cockrell AE, Du Plessis SS. Ultraviolet Light Induced Generation of Reactive Oxygen Species. Advances in Experimental Medicine and Biology. 2017;996:15-23.
35. Shah AA, Sinha AA. Oxidative stress and autoimmune skin disease. European Journal of Dermatology. 2013 Jan 1;23(1):5-13.
36. Shah AA, Dey-Rao R, Seiffert-Sinha K, Sinha AA. Increased oxidative stress in pemphigus vulgaris is related to disease activity and HLA-association. Autoimmunity. 2016 May 18;49(4):248-57.
37. Yesilova Y, Ucmak D, Selek S, Dertlioglu SB, Sula B, Bozkus F, Turan E. Oxidative stress index may play a key role in patients with pemphigus vulgaris. Journal of the European Academy of Dermatology and Venereology. 2013 Apr;27(4):465-7.
38. Baek J, Lee MG. Oxidative stress and antioxidant strategies in dermatology. Redox Report. 2016 Mar 31.
39. Batinac T, Zamolo G, Hadžisejdic I, Žauhar G, Brumini G, Ružic A, et al. Expression of Bcl-2 family proteins in psoriasis. Croatian Medical Journal. 2007 Jun 20;48(3.):319-26.
40. Zamolo G, Coklo M, Duševic DS, Kaštelan M, Batinac T, Materljan E, et al. Expression of p53 and apoptosis in discoid lupus erythematosus. Croatian Medical Journal. 2005 Aug 1;46(4):678-684.
41. Wang X, Bregegere F, Frušic-Zlotkin M, Feinmesser M, Michel B, Milner Y. Possible apoptotic mechanism in epidermal cell acantholysis induced by pemphigus vulgaris autoimmunoglobulins. Apoptosis. 2004 Mar 1;9(2):131- 43.
42. Baroni A, Buommino E, Paoletti I, Orlando M, Ruocco E, Ruocco V. Pemphigus serum and captopril induce heat shock protein 70 and inducible nitric oxide synthase overexpression, triggering apoptosis in human keratinocytes. British Journal of Dermatology. 2004 Jun;150(6):1070-80.
43. Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nature Reviews Cancer. 2002 Aug;2(8):594-604.
44. El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993 Nov 19;75(4):817-25.
45. El-Deiry WS, Harper JW, O’Connor PM, Velculescu VE, Canman CE, Jackman J, et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Research. 1994 Mar 1;54(5):1169-74.
46. Di Leonardo A, Linke SP, Clarkin K, Wahl GM. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes & Development. 1994 Nov 1;8(21):2540-51.
47. Toshiyuki M, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995 Jan 27;80(2):293-9.
48. Geng Y, Walls KC, Ghosh AP, Akhtar RS, Klocke BJ, Roth KA. Cytoplasmic p53 and activated Bax regulate p53- dependent, transcription-independent neural precursor cell apoptosis. Journal of Histochemistry & Cytochemistry. 2010 Mar;58(3):265-75.
49. Ashcroft M, Vousden KH. Regulation of p53 stability. Oncogene. 1999 Dec;18(53):7637-43.
50. Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997 May;387(6630):299- 303.
51. Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harbor Perspectives in Biology 5: a012716.
52. Menon V, Povirk L. Involvement of p53 in the repair of DNA double strand breaks: multifaceted Roles of p53 in homologous recombination repair (HRR) and non-homologous end joining (NHEJ). Subcellular Biochemistry. 2014;85:321-336.
53. Ward IM, Minn K, Jorda KG, Chen J. Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. Journal of Biological Chemistry. 2003 May 30;278(22):19579-82.
54. Venugopal SS, Murrell DF. Diagnosis and Clinical Features of Pemphigus Vulgaris. Immunology and Allergy Clinics of North America. 2012 May;32(2):233-43.
55. Porro AM, Seque CA, Ferreira MC. Pemphigus vulgaris. Anais Brasileiros de Dermatologia. 2019 May;94(3):264-78.
56. Vodo D, Sarig O, Sprecher E. The genetics of pemphigus vulgaris. Frontiers in Medicine. 2018 Aug 14;5:226.
57. Vielmuth F, Waschke J, Spindler V. Loss of desmoglein binding is not sufficient for keratinocyte dissociation in pemphigus. Journal of Investigative Dermatology. 2015 Dec 1;135(12):3068-77.
58. Spindler V, Waschke J. Pemphigus—a disease of desmosome dysfunction caused by multiple mechanisms. Frontiers in Immunology. 2018 Feb 1;9:136.
59. Egu DT, Kugelmann D, Waschke J. Role of PKC and ERK Signaling in Epidermal Blistering and Desmosome Regulation in Pemphigus. Frontiers in Immunology. 2019;10:2883.
60. Spindler V, Waschke J. Desmosomal cadherins and signaling: lessons from autoimmune disease. Cell Communication & Adhesion. 2014 Feb 1;21(1):77-84.
61. Kugelmann D, Rötzer V, Walter E, Egu DT, Fuchs MT, Vielmuth F, et al. Role of Src and cortactin in pemphigus skin blistering. Frontiers in Immunology. 2019 Apr 4;10:626.
62. Sajda T, Sinha AA. Autoantibody signaling in pemphigus vulgaris: development of an integrated model. Frontiers in Immunology. 2018 Apr 19;9:692.
63. Williamson L, Raess NA, Caldelari R, Zakher A, de Bruin A, Posthaus H, Bolli R, Hunziker T, Suter MM, Müller EJ. Pemphigus vulgaris identifies plakoglobin as key suppressor of c-Myc in the skin. The EMBO Journal. 2006 Jul 26;25(14):3298-309.
64. Deyhimi P, Tavakoli P. Study of apoptosis in oral pemphigus vulgaris using immunohistochemical marker Bax and TUNEL technique. Journal of Oral Pathology & Medicine. 2013 May;42(5):409-14.
65. Pelacho B, Natal C, España A, Sanchez-Carpintero I, Iraburu MJ, López-Zabalza MJ. Pemphigus vulgaris autoantibodies induce apoptosis in HaCaT keratinocytes. FEBS Letters. 2004 May 21;566(1-3):6-10.
66. Arredondo J, Chernyavsky AI, Karaouni A, Grando SA. Novel mechanisms of target cell death and survival and of therapeutic action of IVIg in pemphigus. The American Journal of Pathology. 2005 Dec 1;167(6):1531-44.
67. Pacheco-Tovar MG, Avalos-Díaz E, Vega-Memije E, Bollain-y-Goytia JJ, López-Robles E, Hojyo-Tomoka MT, et al. The final destiny of acantholytic cells in pemphigus is Fas mediated. Journal of the European Academy of Dermatology and Venereology. 2009 Jun;23(6):697-701.
68. Puviani M, Marconi A, Pincelli C, Cozzani E. Fas ligand in pemphigus sera induces keratinocyte apoptosis through the activation of caspase-8. Journal of Investigative Dermatology. 2003 Jan 1;120(1):164-7.
69. Sanath AK, Devy AS, Aithal S, Kumar GS, Prasad BG, Pradeep PS. Caspase cascade pathways of apoptosis in oral pemphigus: An immunohistochemical study. Journal of Oral and Maxillofacial Pathology: JOMFP. 2018 Jan;22(1):48.
70. Lanza A, Cirillo N, Femiano F, Gombos F. How does acantholysis occur in pemphigus vulgaris: a critical review. Journal of Cutaneous Pathology. 2006 Jun;33(6):401-12.
71. Grando SA, Bystryn JC, Chernyavsky AI, Frušic- Zlotkin M, Gniadecki R, Lotti R, Milner Y, Pittelkow MR, Pincelli C. Apoptolysis: a novel mechanism of skin blistering in pemphigus vulgaris linking the apoptotic pathways to basal cell shrinkage and suprabasal acantholysis. Experimental Dermatology. 2009 Sep;18(9):764-70.
72. Marchenko S, Chernyavsky AI, Arredondo J, Gindi V, Grando SA. Antimitochondrial Autoantibodies in Pemphigus Vulgaris A Missing Link in Disease Pathophysiology. Journal of Biological Chemistry. 2010 Feb 5;285(6):3695-704.
73. Robles SJ, Adami GR. Agents that cause DNA double strand breaks lead to p16 INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene. 1998 Mar;16(9):1113-23.
74. Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, Koyasu S, Nishikawa T, Amagai M. Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. The Journal of Immunology. 2003 Feb 15;170(4):2170-8.
75. Calkins CC, Setzer SV, Jennings JM, Summers S, Tsunoda K, Amagai M, et al. Desmoglein endocytosis and desmosome disassembly are coordinated responses to pemphigus autoantibodies. Journal of Biological Chemistry. 2006 Mar 17;281(11):7623-34.
76. Luyet C, Schulze K, Sayar BS, Howald D, Müller EJ, Galichet A. Preclinical studies identify non-apoptotic low-level caspase-3 as therapeutic target in pemphigus vulgaris. PLoS One. 2015 Mar 6;10(3):e0119809.
77. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011 Jun;474(7350):179- 83.
78. Aqeilan RI. Hippo signaling: to die or not to die. Cell Death & Differentiation. 2013 Oct;20(10):1287-8.
79. Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiological Reviews. 2014 Oct;94(4):1287-312.
80. Panciera T, Azzolin L, Cordenonsi M, Piccolo S. Mechanobiology of YAP and TAZ in physiology and disease. Nature Reviews Molecular Cell Biology. 2017 Dec;18(12):758-70.
81. Yuan F, Wang J, Li R, Zhao X, Zhang Y, Liu B, et al. A new regulatory mechanism between P53 And YAP crosstalk By SIRT1 mediated deacetylation to regulate cell cycle and apoptosis in A549 cell lines. Cancer Management and Research. 2019;11:8619-8633.
82. Raj N, Bam R. Reciprocal Crosstalk Between YAP1/Hippo Pathway and the p53 Family Proteins: Mechanisms and Outcomes in Cancer. Frontiers in Cell and Developmental Biology. 2019;7:159.
83. Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017 Sep 7;170(6):1062-78.
84. Henseleit U, Zhang J, Wanner R, Haase I, Kolde G, Rosenbach T. Role of p53 in UVB-induced apoptosis in human HaCaT keratinocytes. Journal of Investigative Dermatology. 1997 Dec 1;109(6):722-7.
85. Li X, Ahmad US, Huang Y, Uttagomol J, Rehman A, Zhou K, Warnes G, McArthur S, Parkinson EK, Wan H. Desmoglein-3 acts as a pro-survival protein by suppressing reactive oxygen species and doming whilst augmenting the tight junctions in MDCK cells. Mechanisms of Ageing and Development. 2019 Dec 1;184:111174.
86. Molchadsky A, Rivlin N, Brosh R, Rotter V, Sarig R. p53 is balancing development, differentiation and dedifferentiation to assure cancer prevention. Carcinogenesis. 2010 Sep 1;31(9):1501-8.
87. Saifudeen Z, Dipp S, El-Dahr SS. A role for p53 in terminal epithelial cell differentiation. The Journal of Clinical Investigation. 2002 Apr 15;109(8):1021-30.
88. Johnson JL, Koetsier JL, Sirico A, Agidi AT, Antonini D, Missero C, et al. The desmosomal protein desmoglein 1 aids recovery of epidermal differentiation after acute UV light exposure. Journal of Investigative Dermatology. 2014 Aug 1;134(8):2154-62.
89. Hong AW, Meng Z, Yuan HX, Plouffe SW, Moon S, Kim W, et al. Osmotic stress-induced phosphorylation by NLK at Ser128 activates YAP. EMBO Reports. 2017 Jan;18(1):72-86.
90. Moon S, Kim W, Kim S, Kim Y, Song Y, Bilousov O, et al. Phosphorylation by NLK inhibits YAP-14-3-3- interactions and induces its nuclear localization. EMBO Reports. 2017 Jan;18(1):61-71.
91. Shaw PH. The role of p53 in cell cycle regulation. Pathology-Research and Practice. 1996 Jan 1;192(7):669- 75.
92. Almog N, Rotter V. Involvement of p53 in cell differentiation and development. Biochimica et Biophysica Acta (BBA)/Reviews on Cancer. 1997;1333(1):F1-27.
93. Basu A, Haldar S. The relationship between BcI2, Bax and p53: consequences for cell cycle progression and cell death. Molecular Human Reproduction. 1998 Dec 1;4(12):1099-109.
94. Zeng X, Keller D, Wu L, Lu H. UV but not γ irradiation accelerates p53-induced apoptosis of teratocarcinoma cells by repressing MDM2 transcription. Cancer Research. 2000 Nov 1;60(21):6184-8.
95. Baptiste N, Prives C. p53 in the cytoplasm: a question of overkill?. Cell. 2004 Feb 20;116(4):487-9.
96. Yoshida K, Miki Y. The cell death machinery governed by the p53 tumor suppressor in response to DNA damage. Cancer Science. 2010 Apr;101(4):831-5.