Abstract
Signal transducer and activator of transcription (STAT) 3 plays a key role not only in regulating a variety of biological properties, including survival, proliferation, and metastasis of cancer cells, but also in modifying the tumor microenvironment to promote angiogenesis and immunosuppression, rendering STAT3 a valuable target in cancer. In this review, we discuss the rationale for targeting STAT3 by presenting a succinct overview of STAT3 signaling, outlining an update concerning approaches to directly or indirectly targeting the role of this transcription factor, and highlighting challenges facing therapeutic strategies for inhibiting STAT3 in malignancies. Finally, we also discuss several state-of-the-art approaches for targeting oncogenic STAT3 signaling.
Keywords
Cancer, STAT3, Signaling, Immunotherapy, Combination, YHO-1701
Introduction
Due to its importance in a wide range of cellular processes, including cell proliferation, apoptosis, and immune evasion, the signal transducer and activator of transcription (STAT) 3 signaling pathway has been studied intensively over the past few decades. Dysregulation of the STAT3 signaling pathway is closely associated with initiation and development of various types of hematologic or solid malignancies (approximately 70% of those appearing in humans) [1].
Aberration in upstream activators of the STAT3 signaling pathway, including non-receptor tyrosine kinases (such as breakpoint cluster region-abelson [Bcr-Abl]), Src, and receptor tyrosine kinases (such as epidermal growth factor receptor [EGFR], c-MET, and platelet-derived growth factor [PDGF] receptor), can lead to hyperactivation of STAT3, which serves as an important signaling node for neoplastic cells and the tumor microenvironment (TME). The TME consists not only of tumor cells, but also a variety of host cells, such as immune cells and cancer-associated fibroblasts [2]. The complexity of blocking this pathway can be explained by the presence of other STAT members (STATs 1, 2, 3, 4, 5a, 5b, and 6). Despite the fact that STAT3 shares sequence and structural homology with other STAT family members, the members differ in their intracellular functions, leading to a highly intricate signaling network. For example, STAT1 and STAT3 have great sequence similarity and behave as transcription factors, but under many physiological conditions, they are reciprocally regulated to play opposing roles in cell proliferation and apoptotic cell death [3]. In striking contrast to STAT3 as an oncogene, activated STAT1 functions as a tumor suppressor [3]. Importantly, STAT5, which shows little sequence similarity with STAT3, has also been implicated as an oncogene, primarily in hematopoietic malignancies [4]. In addition, dysregulated STAT3 activation has often been associated with resistance against clinically available molecular-targeted agents [5]. These features make STAT3 an attractive target for cancer therapy, which has motivated the development of targeted approaches [6-9].
A series of targeted therapeutic agents have been developed that have shown promising results against intractable cancers. Examples include EGFR kinase inhibitors (gefitinib, osimertinib, erlotinib, and afatinib) for the treatment of EGFR-mutated non-small cell lung carcinoma (NSCLC), Bcr-Abl inhibitors (imatinib and dasatinib) against leukemia harboring the Bcr- Abl fusion oncogene, and anaplastic lymphoma kinase (ALK) inhibitors (crizotinib, alectinib, and ceritinib) for the treatment of patients with echinoderm microtubuleassociated protein-like 4 (EML4)-ALK positive NSCLC. Unfortunately, however, such inhibition can activate alternative signaling pathways that may later bypass the signaling blockade created by the drug, which in turn leads most patients to succumb to disease progression. Increasing evidence supports compensatory activation of the STAT3 signaling pathway, which limits the therapeutic effects of targeted drugs. For example, the limited therapeutic effect of EGFR inhibitors may be due to STAT3 signaling, a downstream salvage pathway [5,10]. Constitutive activation of STAT3, a downstream point of convergence, can serve as a potential survival pathway against EGFR blockade [5,10]. The limitations of targeted inhibitors as monotherapy emphasize the need for alternative strategies. One possible approach to overcome compensatory activation of survival signaling cascades may be combination therapies, as recently demonstrated by our group and others by simultaneously tackling STAT3 and other signaling pathways [6-9].
In this review, we examine a number of studies and present an update on the role of STAT3 as a therapeutic target, along with current challenges yet to be overcome, and some approaches with high potential.
STAT3 as a Target in Cancer Therapy
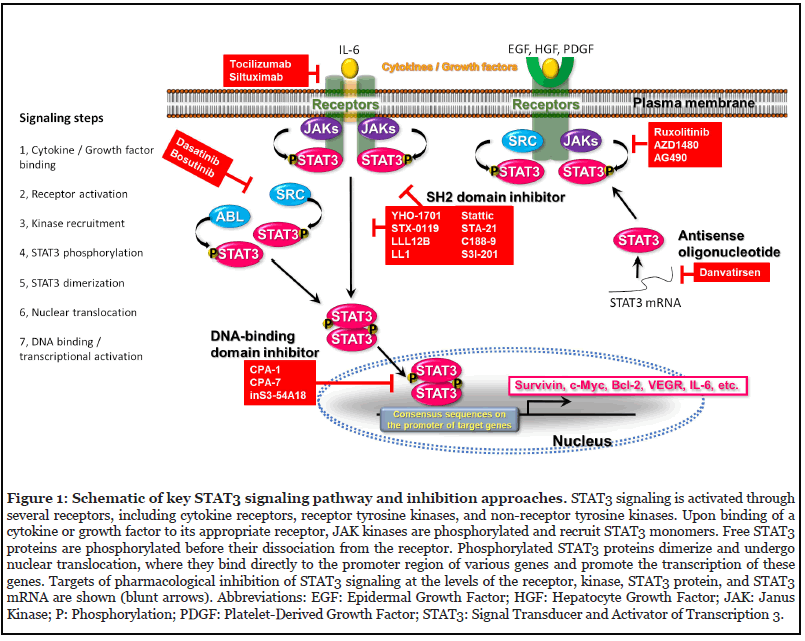
Overactivation of STAT3 is a common molecular characteristic of human malignancies [1,2,6]. When tyrosine kinases such as Janus kinases (JAKs) are activated by stimulation of various cytokines and growth factors, STAT3 is activated by phosphorylation at the Tyr705 residue. Tyrosine-phosphorylated STAT3 induces dimerization through interaction of the Src homology 2 (SH2) domain, translocates into the nucleus and binds to specific DNA sequences to promote the transcription of target genes (Figure 1) [6-9]. Activated STAT3 regulates gene expression involved in survival (e.g., survivin, c-Myc, and Bcl-2) and invasion (e.g., matrix metalloproteinases) of cancer cells, as well as angiogenesis (e.g., vascular endothelial growth factor) and immune escape (e.g., interleukin [IL]-6, IL-10, and TGF-β) within the TME [1,2,6]. STAT3 is constitutively activated in hematologic and solid tumors, while its activation remains transient in normal cells [11], suggesting that STAT3 inhibition is a promising approach for controlling cancers.
Interestingly, it has been observed that only Stat3, but not other family members, knocked out in mice causes embryonic lethality, demonstrating an essential role for this factor in development [12]. Additionally, Stat3 knockout abrogated pluripotency maintenance of embryonic stem cells [13-15], suggesting that aberrant activation of STAT3 can exacerbate the survival of cancer stem cells.
Monotherapy with STAT3 Inhibitors in Cancers
Direct inhibitors of STAT3 signaling
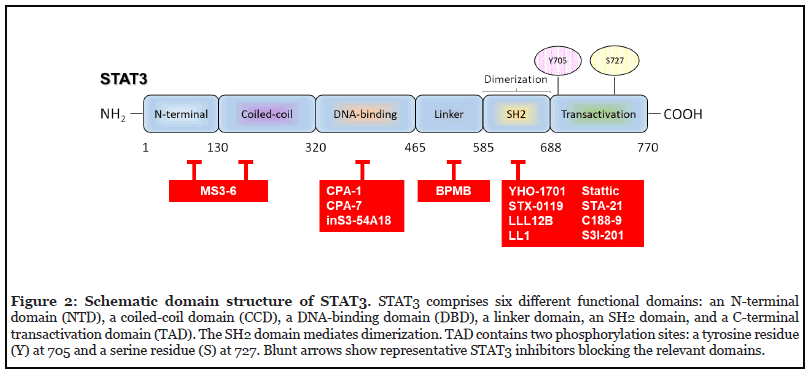
Currently, a common method of blocking the STAT3 pathway is the direct inhibition of STAT3. Direct inhibitors target the SH2 (such as STX-0119, YHO-1701, STA-21, LLL12B, LL1, stattic, S3I-201), DNA-binding (e.g., CPA- 7 and inS3-54A18), coiled-coil, or N-terminal (including MS3-6) domains of STAT3 (Figure 2). Although peptides and peptide mimetics have been shown to disturb the transcriptional activity of STAT3 through interaction with the SH2 domain, some key challenges, including poor membrane permeability and stability, have not yet been sufficiently addressed [16,17]. Although targeted inhibition of STAT3 with decoy oligonucleotides (decoy ODNs) or small interfering RNAs (siRNAs) seems to be attractive methods to specifically abrogate activated STAT3, they also have drawbacks, such as low biomembrane permeability and relative instability [18]. To date, various efforts have been focused on antagonizing the STAT3 signaling pathway [19,20], and some compounds have been investigated both in preclinical and clinical studies (Tables 1 and 2).
| Compound | Cancer | Animal model | Efficacy | Ref. |
|---|---|---|---|---|
| STX-0119 | Lymphoma, Glioblastoma | Xenograft: SCC3 (lymphoma), GBM-SC, U87MG (temozolomide-resistant) | Tumor growth inhibition; Increased life span | [26,27,109] |
| YHO-1701 | HNSCC, NSCLC | Xenograft: SAS (HNSCC), NCI-H2228 (EML4-ALK-expressing NSCLC) |
Tumor growth inhibition | [6,7] |
| LLL12B | Medulloblastoma | Xenograft: D283 | Tumor growth inhibition | [8] |
| LL1 | NSCLC, Colon cancer | Xenograft: A549GR (gefitinib-resistant NSCLC), HCT116 (Colon cancer) |
Tumor growth inhibition | [33,34] |
| Stattic | HNSCC, TNBC | Xenograft: UM-SCC-17B (HNSCC), HCC1806 (TNBC) | Tumor growth inhibition | [29,110] |
| Stattic | HNSCC, TNBC | Xenograft: UM-SCC-17B (HNSCC), HCC1806 (TNBC) | Tumor growth inhibition | [29,110] |
| C188-9 | HNSCC | Xenograft: UM-SCC-17B | Tumor growth inhibition | [30] |
| S3I-201 | Liver cancer | Xenograft: human liver cancer cells | Tumor growth inhibition | [111] |
| CPA-7 | Prostate cancer | Allograft: RM9 | Tumor growth inhibition; Increased life span; Enhanced antitumor immunity |
[112] |
| inS3-54A18 | NSCLC | Xenograft: A549 | Tumor growth inhibition; Anti-metastasis | [43] |
| SD-36 | AML, ALCL | Xenograft: MOLM-16 (AML), SU-DHL-1 (ALCL), SUP-M2 (ALCL) | Tumor growth inhibition | [9] |
| Danvatirsen | ALCL, Vulvar can- cer, NSCLC, MDS/ AML | Xenograft: SUP-M2 (ALCL), A431 (Vulvar cancer), PC-9 (NSCLC), PDX: primary MDS/AML |
Decreased tumor burden; Tumor growth inhibition | [56,113] |
| Napabucacin | DLBCL | Xenograft: SU-DHL-6 | Tumor growth inhibition | [114] |
| OPB-31121 | AML, Stomach cancer | Xenograft: U937 (CALM-AF10- expressing/p53-mutated AML), SNU- 484 (Stomach cancer) | Tumor growth inhibition | [69,70] |
Table 1: In vivo studies of STAT3 inhibitors.
| Drug | Combination | Cancer type | Phase | Identifier |
|---|---|---|---|---|
| Danvatirsen | AZD-5069, durvalumab | HNSCC | I/II | NCT02499328 |
| Durvalumab | Pancreatic cancer, NSCLC | II | NCT02983578 | |
| Cediranib, olaparib, durvalumab, ceralasertib, oleclumab, trastuzumab deruxtecan | Metastatic NSCLC | II | NCT03334617 | |
| Cisplatin, paclitaxel, gemcitabine, carboplatin, fluorouracil, nab- paclitaxel, durvalumab | Solid cancers, NSCLC | I/II | NCT03421353 | |
| Ceralasertib, acalabrutinib | NHL | I/II | NCT03527147 | |
| Cisplatin, gemcitabine, carboplatin, pemetrexed, nab-paclitaxel, durvalumab, oleclumab | Metastatic NSCLC | II | NCT03819465 | |
| Napabucasin | Paclitaxel | Metastatic NSCLC | III | NCT02826161 |
| Irinotecan, fluorouracil, folinic acid | Metastatic CRC | III | NCT03522649 | |
| OPB-51602 | No | Solid cancers | I | NCT01184807 |
| OPB-111077 | Decitabine, venetoclax | AML | I | NCT03063944 |
| No | Cancer, NSCLC, thyroid cancer | II | NCT03158324 | |
| No | AML | I | NCT03197714 | |
| C188-9 | No | Metastatic cancers | I | NCT03195699 |
Abbreviations: ALCL: Anaplastic Large-Cell Lymphoma; AML: Acute Myeloid Leukemia; DLBCL: Diffuse Large B-cell Lymphoma; HNSCC: Head and Neck Squamous Cell Carcinoma; GBM-SC: Glioblastoma Multiforme Stem-like Cells; MDS: Myelodysplastic Syndromes; NSCLC: Non-Small Cell Lung Carcinoma; PDX: Patient-Derived Xenograft; TNBC: Triple-Negative Breast Cancer.
Table 2: STAT3 inhibitors in clinical trials.
Inhibitors targeting the SH2 domain: The STAT3 SH2 domain is one of the most researched targets and is considered an attractive therapeutic target for STAT3 inhibitors because of its central role in multiple key processes, such as tyrosine residue interaction and STAT3:STAT3 dimerization [21], as well as the existence of three subpockets that can be targeted by smallmolecule inhibitors [22]. Most of the small molecules targeting the SH2 domain were identified by structurebased high-throughput virtual screening [6,8,23-25]. For example, STX-0119, an N-[2-(1,3,4-oxadiazolyl)]-4- quinolinecarboxamide derivative, disrupts the binding of STAT3-SH2 [23], thereby inhibiting multi-step events in cancer cells [26]. Oral administration of STX-0119 exerted significant antitumor efficacy in a xenograft model of human lymphoma SCC3 [26], suppressing levels of c-Myc, survivin, cyclin D1, Ki-67, and phospho-STAT3 within the tumors, as well as significant efficacy in xenograft models using glioblastoma multiforme stem-like cells (GBM-SC) from recurrent GBM patients [27]. However, concerns have been raised that poor bioavailability (BA) may limit the utility of this agent, despite its moderate potency [28].
Our group recently discovered that YHO-1701, a structurally optimized analog of STX-0119, has the potential to be a more attractive STAT3 inhibitor than its predecessor, with a BA of 46% [6], which is comparable to that of LLL12B, a structurally optimized analog of STA- 21 [8]. Molecular docking studies with STAT3 suggest a more stable interaction of YHO-1701 with the SH2 domain, corroborating the fact that YHO-1701 exhibits approximately 10-fold stronger activity than STX-0119 in abrogating the STAT3 signaling pathway in the human head and neck squamous cell carcinoma (HNSCC) cell line SAS, which is known to produce IL-6 [6]. Moreover, YHO- 1701, a novel quinolinecarboxamide derivative generated from STX-0119, not only disrupts the level of dimer formation of endogenous STAT3 more clearly than stattic at 10 μM [6], but also inhibits survivin (a downstream target) and proliferation of a diverse range of cancer cells. Furthermore, YHO-1701 exhibits antitumor effects in xenograft models of SAS and NCI-H2228 (NSCLC with EML4-ALK fusion) with repeated oral administration [6,7].
Other compounds, including stattic, S3I-201, C188-9, LLL12, LLL12B, and LL1 similarly abrogated multiple intracellular events, such as the phosphorylation and dimerization of STAT3 through their interruption of the SH2 domain, and inhibited proliferation of cancer cells as well as growth of human tumor xenografts in mice [8,21,29-33]. It is notable that LL1 was highly selective and comparably efficacious compared with napabucasin (BBI608) (described later) [34]. Furthermore, although inhibitors like stattic and S3I-201 are reported to inhibit STAT1 phosphorylation and nuclear translocation [35,36], YHO-1701 also exhibited a weaker inhibitory effect for STAT5 and STAT6 than for STAT3, and little or no effect toward STAT1 and Grb2 in a cell-free assay, indicating higher specificity for STAT3 [6].
Other strategies to disturb the STAT3 SH2 domain are also being examined. Peptides and peptide mimetics are generally designed based on the structure of amino acid residues in the STAT3 protein. PY*LKTK (where Y* indicates p-Tyr705) is derived from the binding peptide sequence of the STAT3-SH2 domain, which binds to the SH2 domain and thereby prevents dimerization [37]. ISS610, a peptidomimetic analog of the tripeptide PY*L, was shown to disrupt STAT3-DNA binding activity, but still had weak intracellular inhibitory properties (IC50 = 1 mM), possibly due to its low intracellular level [38]. S3I-M2001, an oxazole derivative of ISS-610, inhibited the growth of human breast tumor xenografts [39]. PM-73G, another peptidomimetic developed from Y*LPQTV, is also active and efficacious in vivo against the growth of MDA-MB-465 human breast tumor xenografts via intraperitoneal injection [40]. However, some concerns have been raised regarding PM-73G, namely that the authors of studies on PM-73G reported no observed changes in the expression of either cyclin D1 or survivin, which are known downstream targets regulated by activated STAT3, and that inhibition of proliferation in cell lines, which all harbor constitutively active STAT3, occurred only at higher concentrations than those required to inhibit STAT3 phosphorylation at Tyr705, which is suggestive of STAT3-independent effects [40,41]. Despite substantial research into peptide inhibitors of STAT3, poor membrane permeability and lack of stability in vivo are key examples of the problems that have impeded their further clinical development as therapeutics [38].
The small-molecule proteolysis-targeting chimera (PROTAC)-based strategy has attracted increasing attention because it not only can obstruct target protein function, but also can counteract increased target protein expression [42]. SD-36, a novel PROTAC targeting STAT3 protein, demonstrated high selectivity over other STAT proteins [9]. SD-36 consists of the cell-permeable STAT3- SH2 domain-targeting inhibitor, SI-109, a linker, and an analog of the CRBN ligand lenalidomide for E3 ubiquitin ligase [9]. Moreover, SD-36 treatment achieved complete and long-lasting tumor regression in multiple xenograft mouse models of leukemia and lymphoma cell lines (MOLM-16, SU-DHL-1, and SUP-M2) without exhibiting obvious signs of toxicity [9], suggesting that PROTAC STAT3 degraders may be reliable in development of therapeutics. Considering that STAT3 signaling plays a pivotal role in numerous biological processes in the development of both solid and hematologic malignancies, it is also worth considering examining the effectiveness of SD-36 in solid tumors in the future.
Inhibitors targeting the DNA-binding domain (DBD): Recent studies have demonstrated that multiple STAT3 DNA-binding inhibitors exert biological activity both in vitro and in vivo [43,44]. InS3-54 (designed by an improved in silico approach) selectively inhibits STAT3 binding to DNA without affecting the activation and dimerization of STAT3 [43,44]. Notably, inS3-54A18, an optimized analog of inS3-54, not only inhibits STAT3 DNA binding and gene expression of STAT3 [43], but also significantly inhibited downstream target genes of STAT3 [43-45] and decreased growth and metastasis in an NSCLC xenograft model of A549 following repeated oral administration [43]. These findings suggest that inS3- 54A18 may be an attractive STAT3 inhibitor because it directly blocks the expression of pro-oncogenes.
Decoy ODNs attach to the STAT3-DBD and block its binding with the responsive elements, consequently suppressing gene expression [46]. Specific ODN sequences have thus far been tested: intratumoral injection of ODN 5’-CATTTCCCGTAAATC-3’ downregulated STAT3 target gene expression and suppressed tumor growth in a U251 glioblastoma xenograft model [47]. Another study revealed that when 3.2 mg/kg of a STAT3 double-stranded ODN (dsODN) decoy was injected intramuscularly in monkeys, no apparent adverse event was observed, despite downregulated gene expression of Bcl-XL and cyclin D1 at the injection site [48]. A more stable cyclic version of the decoy, 5’-CATTTCCCGTAAATC-3’, which is resistant to serum nucleases, has also been shown to suppress HNSCC tumor growth and expression of STAT3 target genes in tumors [49]. An experimental study was carried out to evaluate the biological effects of STAT3 dsODN decoy when injected intratumorally in HNSCC patients (NCT00696176). This study indicates inhibition of genes targeted by STAT3, yet the decoy degrades rapidly in serum, indicating that although the STAT3 ODN decoy approach is promising, ODNs are not amenable to systemic delivery [49,50].
Although studies have also confirmed the efficacy of other STAT3 DBD inhibitors, including platinum (IV) compounds such as CPA-1, CPA-7, and IS3 295 [51,52], the use of these as specific STAT3 inhibitors might be limited, due in large part to the lack of specificity to STAT3.
Inhibitors targeting STAT3 mRNA: Antisense oligonucleotides (ASOs) can inhibit STAT3 expression and function by downregulating STAT3 messenger RNA (mRNA) [53,54]. For example, danvatirsen, also known as AZD9150 or ISIS 481464, has been shown to reduce STAT3 mRNA and protein levels and block cell proliferation. Additionally, although danvatirsen is unsuitable for systemic administration because of its rapid degradation, similar to other siRNAs and decoy ODNs [55], the agent diminished the STAT3 protein level, thereby inhibiting the growth of subcutaneous xenograft tumors derived from human hepatocellular carcinoma and diffuse large B-cell lymphoma via intradermal and intratumoral injections, respectively [53,54,56], and it was well tolerated at doses up to 30 mg/kg/week for 6 weeks in monkeys [57]. Critically, tumor biopsy samples from danvatirsen-treated patients have shown that its uptake was observed mainly in cells of the TME (but not cancer cells), suggesting that the clinical benefit was not due to direct tumor cell modulation, but rather through remodeling of the suppressive TME [58]. Taken together, these results indicate that although STAT3 ASOs provide high specificity and potency, rapid degradation is the main problem that could impede their clinical utility. However, the biopsy data of danvatirsen provide a promising rationale for testing its combination with immunotherapy in the clinic [58].
Miscellaneous STAT3 inhibitors: Napabucasin has been reported as a STAT3 inhibitor; however, its exact mechanism of action remains controversial. Some claim that napabucasin inhibits STAT3 by binding to its SH2 domain and preventing its dimerization and subsequent activation [59,60], but others claim that it binds to the DBD [2] or in a pocket between the linker and the DBD of STAT3 [61]. Napabucasin has been reported to act on multiple other oncogenic pathways, including the WNT/β-catenin pathway, and it is known as a STAT3 and cancer cell stemness inhibitor [62-66]. Previous studies have shown that napabucasin abrogates the relapse and metastasis of tumor xenografts by inhibiting cancer stem cells [65,67]. However, a phase III study [68] revealed that addition of napabucasin to the FOLFIRI regimen (irinotecan, fluorouracil, and folinic acid) with or without bevacizumab did not improve the overall survival of patients with metastatic colorectal cancer (mCRC), failing to meet the primary endpoint.
It has been demonstrated that OPB-31121 and -51602 constitute biological responses in vitro and in vivo with varying potency against diverse tumor models harboring constitutively active STAT3 [69,70]. OPB-31121, OPB- 51602, and OPB-111077 have already been tested in clinical trials. Unfortunately, although OPB-31121 was found to have preliminary antitumor activity in patients with advanced solid tumors [71], other studies demonstrated that it has dose-dependent toxicities (grade 3 vomiting and grade 3 diarrhea) without objective responses in patients with advanced solid tumors [72] and has insufficient antitumor activity in HCC patients [73]. A phase I study of OPB-51602 revealed that it demonstrates promising antitumor activity, particularly in NSCLC, but has a poor safety profile for continuous dosing [74]. Although OPB- 111077 was found to be better tolerated, only modest clinical activity was observed [75]. Notably, the exact mechanism of action of OPB compounds in inhibiting STAT3 signaling is not always clear. OPB-31121 and -51602 bind to the SH2 domain; however, molecular docking and dynamic simulations indicated that their binding sites do not overlap with any other STAT3 inhibitors [69,70], reviewed in [76]. Previous reports suggest that OPB- 31121 downregulates JAK2 and gp130 expression and disrupts JAK2 phosphorylation in gastric cancer cells [70], suggesting its function as an upstream inhibitor. Thus, it remains ambiguous whether the antitumor effects produced by OPB compounds are based on direct and indirect blockages of STAT3 signaling, because there is a lack of information on OPB-51602 modes of action (also reviewed in [77]).
Taken together, the aforementioned evidence indicates that although encouraging data were obtained from preclinical studies, the use of OPB compounds as specific STAT3 inhibitors might be limited because of their effects on many signal transduction pathways. Additionally, there have been concerns about the potentially severe toxicities of OPB-51602 and OPB-31121, including susceptibility to opportunistic infections, which may limit their further development [74], reviewed in [55]. However, given the favorable safety profiles of OPB-111077, its combination with other treatment strategies might open a new avenue for future cancer therapy.
Indirect inhibitors of STAT3 signaling
Another competitive approach of blocking the STAT3 pathway is indirectly inhibiting upstream tyrosine kinases. Activation of STAT3 signaling is mediated by receptor tyrosine kinases (including EGFR, c-MET, etc.) and nonreceptor tyrosine kinases such as Src and Abl, along with cytokine receptors associated with JAKs [78] because such upstream activators converge on STAT3 signaling, providing an indirect approach to disrupt the STAT3 signaling pathway (Figure 1). However, inhibitors can also affect other STAT family members simultaneously, indicating a lack of specificity to STAT3 [79]. JAK inhibitors such as ruxolitinib (ICNB18424) and tofacitinib have been approved by the FDA for the treatment of myelofibrosis and rheumatoid arthritis, respectively [80-82]. Moreover, the JAK inhibitor baricitinib (Olumiant), a rheumatoid drug, has recently been approved for treating coronavirus disease 2019 (COVID-19) [83]. JAK inhibitors such as LS- 104, AG490, lestaurtinib (CEP701), ruxolitinib, WP1066, and AZD1480 have been tested in tumor xenograft models [84-88]. Results indicate that AG490 downregulates STAT3 levels, reduces STAT3 DNA binding, and abrogates the proliferation of cancer cells by inhibiting upstream JAK2 kinase [89,90]. Ruxolitinib, an oral JAK 1/2 inhibitor, decreases phosphorylation of STAT3 and proliferation of human leukemia HEL92.1.7 cells with the JAK2 V617F mutation. [88]. It has been reported that AZD1480 inhibits JAK1 and 2 kinases, with IC50 values of 1.3 and <0.4 nM, respectively, in enzyme assays [91], and induces antitumor effects in xenograft models including human glioblastoma and NSCLC [85,92]. In addition to its potent activity against JAKs, AZD1480 inhibited the proliferation of multiple Hodgkin lymphoma cells harboring active JAK/ STAT proteins when applied at a higher concentration (5 μM) [91], suggesting that it also induced antitumor effects in a JAK/STAT3-independent manner.
Although some upstream inhibitors have advanced to clinical trials [55,93,94], it has been observed that JAK inhibitors have limitations, including off-target neurological adverse events [55,93]. For instance, earlyphase clinical trials of JAK1/2 inhibitors (e.g., AZD1480) and Src inhibitors (e.g., dasatinib) have revealed limited efficacy or excessive toxicity in cancer patients [93,95]. The JAK2 inhibitor lestaurtinib showed dose-dependent suppression of phosphorylated STAT3, which was associated with grade 3 or 4 adverse events (mainly myelosuppression) in some patients with myelofibrosis [96]. Possible explanations for these adverse events include their influence on multiple downstream signaling pathways. Furthermore, treatment with molecular-targeted drugs may not necessarily result in the downregulation of STAT3 signaling, most likely due to the existence of compensatory mechanisms [5-7]. Although these inhibitors are beneficial in treating diseases, including autoimmune diseases, myeloproliferative disorders, and COVID-19 [79-83,97], targeting one of the upstream effectors of STAT3 is unlikely to be sufficient for cancer therapy.
Combination Strategies with STAT3 Inhibitors in Cancers
STAT3 is a crucial convergence point in several ligand/ receptor pathways and non-receptor tyrosine kinase pathways. The consequent crosstalk among these signaling pathways may alter sensitivity to molecular-targeted drugs, such as Bcr-Abl inhibitors, EGFR inhibitors, and ALK inhibitors [7]. For example, pathway-targeted drug therapies can activate STAT3 signaling in cancer cells driven by diverse activated kinases, including EGFR, HER2, ALK, and c-MET, as well as mutant KRAS [98]. These findings suggest that the compensatory STAT3 activation mechanism of pharmacological inhibition by targeted drugs, together with conventional and immunotherapeutic agents, limits the efficacy of single-drug treatment. In other words, the simultaneous combination of STAT3- targeted drugs with other anticancer therapeutics with different mechanisms of action will likely elicit more salutary antitumor responses.
Recent research in our group using YHO-1701, a STAT3 SH2 domain inhibitor, showed that additive or synergistic effects were observed in approximately two-thirds of the 33 “combination + human cell line” sets. YHO-1701/ sorafenib combination in SAS oral cancer cells (IL-6 signaling) and YHO-1701/Bcr-Abl inhibitors (imatinib, dasatinib) against lymphoma cell lines K562 and SUP-B15 (Bcr-Abl fusion), YHO-1701/ALK inhibitors (crizotinib, alectinib, ceritinib) against NCI-H2228 cells (EML4-ALK fusion), and YHO-1701/osimertinib against NCI-H1975 NSCLC cells (L858R/T790M EGFR double-mutant) are some examples [7]. Notably, crizotinib and alectinib inhibited phospho-ERK, and addition of the STAT3 inhibitor YHO-1701 to these agents resulted in greater downregulation of the STAT3-survivin signaling pathway, as well as promotion of beneficial antiproliferative effects in NCI-H2228 cells, which was in agreement with the results of a previous study emphasizing the importance of dual interruption of the STAT3 and ERK signaling pathways in NCI-H2228 cells [99]. Additionally, addition of YHO- 1701 to alectinib exhibited a significant combination effect against NCI-H2228 xenografts by alleviating the NCI-H2228 xenograft-induced body weight loss with no systemic adverse effects. YHO-1701 also enhanced the antitumor activity of sorafenib in an IL-6-secreting SAS xenograft model [6].
Likewise, a recently published study found that LLL12B in combination with cisplatin, a conventional chemotherapeutic agent, suppressed tumor growth in D283 and D425 medulloblastoma xenografts [8].
Increasing evidence supports the promise of the immunomodulatory effects of STAT3 inhibition. For instance, a preclinical study has shown enhanced survival and antitumor effects by combining anti-CTLA-4 treatment (ipilimumab) with GPB730, a direct STAT3 inhibitor, in a syngeneic prostate cancer mouse model [100]. Another study demonstrated that administration of a highly absorptive form of extracted curcumin targeting STAT3 in combination with an anti-PD-1 or anti-PD-L1 antibody was able to augment the antitumor effect in an MC-38 syngeneic murine model [101], providing a rationale for combination immunotherapies with STAT3 inhibitors. Clinical trials investigating the dual blockade of the STAT3 and PD-1 axes in malignancies are underway (including NCT02499328, NCT02983578, NCT03334617, NCT03421353, and NCT03425006) [28,102,103].
Future Directions and Conclusion
To date, intensive efforts have been made to target STAT3 for the development and application of new drugs; however, there are still no clinically applicable STAT3- selective inhibitors. The preceding discussion highlights examples of the key concerns that must be considered, including toxicity, lack of stability, and specificity.
Interestingly, several groups have shown that a pool of STAT3 translocates into the mitochondria other than the nucleus, and that this is dependent on phospho- Ser727 but independent of Tyr705 phosphorylation [104,105]. Similarly, it has been observed that nuclear translocation and DNA binding of STAT3 can occur independently of tyrosine phosphorylation [106]. These observations indicate that targeting the SH2 domain may not be sufficient to fully abrogate STAT3 oncogenic functions (reviewed in [107]), which may partially hinder the efficacy of such inhibitors. Therefore, novel strategies that target other functional domains must be considered in the future. Several novel approaches have recently emerged and have yielded encouraging results. One recent report has shown that BPMB inhibits the transcriptional activity of STAT3, presumably by inducing stable STAT3 complexes, including a homodimer, through a bifunctional intermolecular covalent reaction with Cys550 in the linker domain, despite its inability to reduce the phosphorylation and nuclear translocation of STAT3. BPMB selectively inhibited the proliferation of human breast cancer cell lines with constitutively activated STAT3 [108]. Another group reported the identification of a novel druggable binding site within the DBD [61], which may contribute to the discovery of attractive STAT3 inhibitors because it indicates that expression of pro-oncogenes can be directly blocked.
Additionally, extensive crosstalk and alternative signaling pathways in cells likely render single-agent STAT3 inhibition less effective. Although most of the direct inhibitors have shown moderate efficacy in treating cancers as single agents, they may act synergistically with clinically available therapies, which will likely elicit enhanced antitumor responses.
In this regard, the combination of STAT3 inhibitors with immunotherapeutic agents may be a beneficial therapeutic approach for cancer patients. Collectively, although more extensive studies are needed, we believe that the dual blockade of STAT3 and rational therapeutic targets is a key concept for improved cancer treatment, given the fact that the STAT3 pathway is inextricably intertwined with different signaling pathways in cells.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work. KT prepared the original draft of manuscript; KT and MT prepared the figures and tables; MT and AA reviewed and edited the manuscript.
References
2. Zou S, Tong Q, Liu B, Huang W, Tian Y, Fu X. Targeting STAT3 in Cancer Immunotherapy. Mol Cancer. 2020;19(1):145.
3. Regis G, Pensa S, Boselli D, Novelli F, Poli V. Ups and downs: The STAT1:STAT3 seesaw of Interferon and gp130 receptor signaling. Semin Cell Dev Biol. 2008;19(4):351-9.
4. Koppikar P, Lui VW, Man D, Xi S, Chai RL, Nelson E, et al. Constitutive Activation of STAT5 Contributes to Tumor Growth, Epithelial-Mesenchymal Transition, and Resistance to EGFR Targeting. Clin Cancer Res. 2008;14(23):7682-90.
5. Wang X, Crowe PJ, Goldstein D, Yang JL. STAT3 inhibition, a novel approach to enhancing targeted therapy in human cancers (Review). Int J Oncol. 2012;41(4):1181- 91.
6. Nishisaka F, Taniguchi K, Tsugane M, Hirata G, Takagi A, Asakawa N, et al. Antitumor activity of a novel oral signal transducer and activator of transcription 3 inhibitor YHO-1701. Cancer Sci. 2020;111(5):1774-84.
7. Taniguchi K, Konishi H, Yoshinaga A, Tsugane M, Takahashi H, Nishisaka F, et al. Efficacy of combination treatment using YHO-1701, an orally active STAT3 inhibitor, with molecular-targeted agents on cancer cell lines. Sci Rep. 2021;11(1):6685.
8. Chen X, Pan L, Wei J, Zhang R, Yang X, Song J, et al. LLL12B, a small molecule STAT3 inhibitor, induces growth arrest, apoptosis, and enhances cisplatinmediated cytotoxicity in medulloblastoma cells. Sci Rep. 2021;11(1):6517.
9. Bai L, Zhou H, Xu R, Zhao Y, Chinnaswamy K, McEachern D, et al. A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell. 2019;36(5):498-511.e17.
10. Lai SY, Johnson FM. Defining the role of the JAKSTAT pathway in head and neck and thoracic malignancies: Implications for future therapeutic approaches. Drug Resist Updat. 2010;13(3):67-78.
11. Zhang HF, Lai R. STAT3 in Cancer-Friend or Foe? Cancers. 2014;6(3):1408-40.
12. Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A. 1997;94(8):3801-4.
13. Raz R, Lee CK, Cannizzaro LA, d’Eustachio P, Levy DE. Essential role of STAT3 for embryonic stem cell pluripotency. Proc Natl Acad Sci U S A. 1999;96(6):2846-51.
14. Zhang Y, Wang D, Xu J, Wang Y, Ma F, Li Z, et al. Stat3 activation is critical for pluripotency maintenance. J Cell Physiol. 2019;234(2):1044-51.
15. Cheng CC, Liao PN, Ho AS, Lim KH, Chang J, Su YW, et al. STAT3 exacerbates survival of cancer stemlike tumorspheres in EGFR-positive colorectal cancers: RNAseq analysis and therapeutic screening. J Biomed Sci. 2018;25(1):60.
16. Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E, et al. Phosphotyrosyl Peptides Block Stat3-mediated DNA Binding Activity, Gene Regulation, and Cell Transformation. J Biol Chem. 2001;276(48):45443-55.
17. Zhang X, Sun Y, Pireddu R, Yang H, Urlam MK, Lawrence HR, et al. A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res. 2013;73(6):1922-33.
18. Pedroso de Lima MC, Simões S, Pires P, Faneca H, Düzgüneş N. Cationic lipid-DNA complexes in gene delivery: from biophysics to biological applications. Adv Drug Deliv Rev. 2001;47(2-3):277-94.
19. Lee DS, O’Keefe RA, Ha PK, Grandis JR, Johnson DE. Biochemical Properties of a Decoy Oligodeoxynucleotide Inhibitor of STAT3 Transcription Factor. Int J Mol Sci. 2018;19(6):1608.
20. Ramasamy T, Chen X, Qin B, Johnson DE, Grandis JR, Villanueva FS. STAT3 decoy oligonucleotide-carrying microbubbles with pulsed ultrasound for enhanced therapeutic effect in head and neck tumors. PLoS One. 2020;15(11):e0242264.
21. Zhang X, Yue P, Fletcher S, Zhao W, Gunning PT, Turkson J. A novel small-molecule disrupts Stat3 SH2 domain-phosphotyrosine interactions and Stat3- dependent tumor processes. Biochem Pharmacol. 2010;79(10):1398-409.
22. Debnath B, Xu S, Neamati N. Small molecule inhibitors of signal transducer and activator of transcription 3 (Stat3) protein. J Med Chem. 2012;55(15):6645-68.
23. Matsuno K, Masuda Y, Uehara Y, Sato H, Muroya A, Takahashi O, et al. Identification of a New Series of STAT3 Inhibitors by Virtual Screening. ACS Med Chem Lett. 2010;1(8):371-5.
24. Yuan J, Zhang F, Niu R. Multiple regulation pathways and pivotal biological functions of STAT3 in cancer. Sci Rep. 2015;5:17663.
25. Xu X, Kasembeli MM, Jiang X, Tweardy BJ, Tweardy DJ. Chemical Probes that Competitively and Selectively Inhibit Stat3 Activation. PLoS One. 2009;4(3):e4783.
26. Ashizawa T, Miyata H, Ishii H, Oshita C, Matsuno K, Masuda Y, et al. Antitumor activity of a novel small molecule STAT3 inhibitor against a human lymphoma cell line with high STAT3 activation. Int J Oncol. 2011;38(5):1245-52.
27. Ashizawa T, Miyata H, Iizuka A, Komiyama M, Oshita C, Kume A, et al. Effect of the STAT3 inhibitor STX-0119 on the proliferation of cancer stem-like cells derived from recurrent glioblastoma. Int J Oncol. 2013;43(1):219-27.
28. Ou A, Ott M, Fang D, Heimberger AB. The Role and Therapeutic Targeting of JAK/STAT Signaling in Glioblastoma. Cancers. 2021;13(3):437.
29. Adachi M, Cui C, Dodge CT, Bhayani MK, Lai SY. Targeting STAT3 inhibits growth and enhances radiosensitivity in head and neck squamous cell carcinoma. Oral Oncol. 2012;48(12):1220-6.
30. Bharadwaj U, Eckols TK, Xu X, Kasembeli MM, Chen Y, Adachi M, et al. Small-molecule inhibition of STAT3 in radioresistant head and neck squamous cell carcinoma. Oncotarget. 2016;7(18):26307-30.
31. Zuo M, Li C, Lin J, Javle M. LLL12, a novel small inhibitor targeting STAT3 for hepatocellular carcinoma therapy. Oncotarget. 2015;6(13):10940-9.
32. Nie Y, Li Y, Hu S. A novel small inhibitor, LLL12, targets STAT3 in non-small cell lung cancer in vitro and in vivo. Oncol Lett. 2018;16(4):5349-54.
33. Liu Z, Ma L, Sun Y, Yu W, Wang X. Targeting STAT3 signaling overcomes gefitinib resistance in non-small cell lung cancer. Cell Death Dis. 2021;12(6):561.
34. Liu Z, Wang H, Guan L, Lai C, Yu W, Lai M. LL1, a novel and highly selective STAT3 inhibitor, displays anti-colorectal cancer activities in vitro and in vivo. Br J Pharmacol. 2020;177(2):298-313.
35. Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13(11):1235-42.
36. Lin L, Benson DM, Jr., DeAngelis S, Bakan CE, Li PK, Li C, et al. A small molecule, LLL12 inhibits constitutive STAT3 and IL-6-induced STAT3 signaling and exhibits potent growth suppressive activity in human multiple myeloma cells. Int J Cancer. 2012;130(6):1459-69.
37. Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E, et al. Phosphotyrosyl peptide block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J Biol Chem. 2001;276(48):45443-55.
38. Turkson J, Kim JS, Zhang S, Yuan J, Huang M, Glenn M, et al. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol Cancer Ther. 2004;3(3):261-9.
39. Siddiquee KA, Gunning PT, Glenn M, Katt WP, Zhang S, Schrock C, et al. An Oxazole-Based Small-Molecule Stat3 Inhibitor Modulates Stat3 Stability and Processing and Induces Antitumor Cell Effects. ACS Chem Biol. 2007;2(12):787-98.
40. Auzenne EJ, Klostergaard J, Mandal PK, Liao WS, Lu Z, Gao F, et al. A phosphopeptide mimetic prodrug targeting the SH2 domain of STAT3 inhibits tumor growth and angiogenesis. J Exp Ther Oncol. 2012;10(2):155-62.
41. Mandal PK, Gao F, Lu Z, Ren Z, Ramesh R, Birtwistle JS, et al. Potent and Selective Phosphopeptide Mimetic Prodrugs Targeted to the Src Homology 2 (SH2) Domain of Signal Transducer and Activator of Transcription 3. J Med Chem. 2011;54(10):3549-63.
42. Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16(2):101-14.
43. Huang W, Dong Z, Chen Y, Wang F, Wang CJ, Peng H, et al. Small-molecule inhibitors targeting the DNA-binding domain of STAT3 suppress tumor growth, metastasis and STAT3 target gene expression in vivo. Oncogene. 2016;35(6):783-92.
44. Huang W, Dong Z, Wang F, Peng H, Liu JY, Zhang JT. A Small Molecule Compound Targeting STAT3 DNABinding Domain Inhibits Cancer Cell Proliferation, Migration, and Invasion. ACS Chem Biol. 2014;9(5):1188- 96.
45. Zhao C, Li H, Lin HJ, Yang S, Lin J, Liang G. Feedback Activation of STAT3 as a Cancer Drug-Resistance Mechanism. Trends Pharmacol Sci. 2016;37(1):47-61.
46. Furqan M, Akinleye A, Mukhi N, Mittal V, Chen Y, Liu D. STAT inhibitors for cancer therapy. J Hematol Oncol. 2013;6:90.
47. Shen J, Li R, Li G. Inhibitory Effects of Decoy-ODN Targeting Activated STAT3 on Human Glioma Growth In Vivo. In Vivo. 2009;23(2):237-43.
48. Sen M, Tosca PJ, Zwayer C, Ryan MJ, Johnson JD, Knostman KA, et al. Lack of toxicity of a STAT3 decoy oligonucleotide. Cancer Chemother Pharmacol. 2009;63(6):983-95.
49. Sen M, Thomas SM, Kim S, Yeh JI, Ferris RL, Johnson JT, et al. First-in-Human Trial of a STAT3 Decoy Oligonucleotide in Head and Neck Tumors: Implications for Cancer Therapy. Cancer Discov. 2012;2(8):694-705.
50. Johnston PA, Grandis JR. STAT3 SIGNALING: Anticancer Strategies and Challenges. Mol Interv. 2011;11(1):18-26.
51. Turkson J, Zhang S, Palmer J, Kay H, Stanko J, Mora LB, et al. Inhibition of constitutive signal transducer and activator of transcription 3 activation by novel platinum complexes with potent antitumor activity. Mol Cancer Ther. 2004;3(12):1533-42.
52. Turkson J, Zhang S, Mora LB, Burns A, Sebti S, Jove R. A Novel Platinum Compound Inhibits Constitutive Stat3 Signaling and Induces Cell Cycle Arrest and Apoptosis of Malignant Cells. J Biol Chem. 2005;280(38):32979-88.
53. Li WC, Ye SL, Sun RX, Liu YK, Tang ZY, Kim Y, et al. Inhibition of growth and metastasis of human hepatocellular carcinoma by antisense oligonucleotide targeting signal transducer and activator of transcription 3. Clin Cancer Res. 2006;12(23):7140-8.
54. Reilley MJ, McCoon P, Cook C, Lyne P, Kurzrock R, Kim Y, et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: results of a phase 1b trial. J Immunother Cancer. 2018;6(1):119.
55. Arshad S, Naveed M, Ullia M, Javed K, Butt A, Khawar M, et al. Targeting STAT-3 signaling pathway in cancer for development of novel drugs: Advancements and challenges. Genet Mol Biol. 2020;43(1):e20180160.
56. Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, et al. AZD9150, a Next-Generation Antisense Oligonucleotide Inhibitor of STAT3 with Early Evidence of Clinical Activity in Lymphoma and Lung Cancer. Sci Transl Med. 2015;7(314):314ra185.
57. Burel SA, Han SR, Lee HS, Norris DA, Lee BS, Machemer T, et al. Preclinical evaluation of the toxicological effects of a novel constrained ethyl modified antisense compound targeting signal transducer and activator of transcription 3 in mice and cynomolgus monkeys. Nucleic Acid Ther. 2013;23(3):213-27.
58. Proia TA, Singh M, Woessner R, Carnevalli L, Bommakanti G, Magiera L, et al. STAT3 Antisense Oligonucleotide Remodels the Suppressive Tumor Microenvironment to Enhance Immune Activation in Combination with Anti-PD-L1. Clin Cancer Res. 2020;26(23):6335-49.
59. Zhou Q, Peng C, Du F, Zhou L, Shi Y, Du Y, et al. Design, synthesis and activity of BBI608 derivatives targeting on stem cells. Eur J Med Chem. 2018;151:39-50.
60. Li C, Chen C, An Q, Yang T, Sang Z, Yang Y, et al. A novel series of napacucasin derivatives as orally active inhibitors of signal transducer and activator of transcription 3 (STAT3). Eur J Med Chem. 2019;162:543-54.
61. Sabanés Zariquiey F, da Souza JV, Estrada-Tejedor R, Bronowska AK. If You Cannot Win Them, Join Them: Understanding New Ways to Target STAT3 by Small Molecules. ACS Omega. 2019;4(9):13913-21.
62. Hanafi M, Chen X, Neamati N. Discovery of a Napabucasin PROTAC as an Effective Degrader of the E3 Ligase ZFP91. J Med Chem. 2021;64(3):1626-48.
63. Beyreis M, Gaisberger M, Jakab M, Neureiter D, Helm K, Ritter M, et al. The Cancer Stem Cell Inhibitor Napabucasin (BBI608) Shows General Cytotoxicity in Biliary Tract Cancer Cells and Reduces Cancer Stem Cell Characteristics. Cancers (Basel). 2019;11(3):276.
64. Xiao L, Niu HJ, Qu TL, Zhang XF, Du FY. Targeting cancer stem cells in drug discovery: Current state and future perspectives. World J Stem Cells. 2019;11(7):398- 420.
65. Li Y, Rogoff HA, Keates S, Gao Y, Murikipudi S, Mikule K, et al. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc Natl Acad Sci U S A. 2015;112(6):1839-44.
66. Zhang Y, Jin Z, Zhou H, Ou X, Xu Y, Li H, et al. Suppression of prostate cancer progression by cancer cell stemness inhibitor napabucasin. Cancer Med. 2016;5(6):1251-8.
67. Beyreis M, Gaisberger M, Jakab M, Neureiter D, Helm K, Ritter M, et al. The Cancer Stem Cell Inhibitor Napabucasin (BBI608) Shows General Cytotoxicity in Biliary Tract Cancer Cells and Reduces Cancer Stem Cell Characteristics. Cancers. 2019;11(3):276.
68. Grothey A, Shah MA, Yoshino T, Van Cutsem E, Taieb J, Xu R, et al. CanStem303C trial: A phase III study of napabucasin (BBI-608) in combination with 5-fluorouracil (5-FU), leucovorin, irinotecan (FOLFIRI) in adult patients with previously treated metastatic colorectal cancer. (mCRC). J Clin Oncol. 2017;35(suppl 15).
69. Hayakawa F, Sugimoto K, Harada Y, Hashimoto N, Ohi N, Kurahashi S, et al.: A novel STAT inhibitor, OPB-31121, has a significant antitumor effect on leukemia with STATaddictive oncokinases. Blood Cancer J. 2013;3(11):e166.
70. Kim MJ, Nam HJ, Kim HP, Han SW, Im SA, Kim TY, et al. OPB-31121, a novel small molecular inhibitor, disrupts the JAK2/STAT3 pathway and exhibits an antitumor activity in gastric cancer cells. Cancer Lett. 2013;335(1):145-52.
71. Oh DY, Lee SH, Han SW, Kim MJ, Kim TM, Kim TY, et al. Phase I study of OPB-31121, an Oral STAT3 inhibitor, in patients with advanced solid tumors. Cancer Res Treat. 2015;47(4):607-15.
72. Bendell JC, Hong DS, Burris HA 3rd, Naing A, Jones SF, Falchook G, et al. Phase 1, open-label, dose-escalation, and pharmacokinetic study of STAT3 inhibitor OPB-31121 in subjects with advanced solid tumors. Cancer Chemother Pharmacol. 2014;74(1):125-30.
73. Okusaka T, Ueno H, Ikeda M, Mitsunaga S, Ozaka M, Ishii H, et al. Phase 1 and pharmacological trial of OPB-31121, a signal transducer and activator of transcription-3 inhibitor in patients with advanced hepatocellular carcinoma. Hepatol Res. 2015;45(13):1283- 91.
74. Wong AL, Soo RA, Tan DS, Lee SC, Lim JS, Marban PC, et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann Oncol. 2015;26(5):998-1005.
75. Tolcher A, Flaherty K, Shapiro GI, Berlin J, Witzig T, Habermann T, et al. A First-in-Human Phase I Study of OPB-111077, a Small-Molecule STAT3 and Oxidative Phosphorylation Inhibitor, in Patients with Advanced Cancers. Oncologist. 2018;23(6):658-e72.
76. Gu Y, Mohammad IS, Liu Z, et al. Overview of the STAT-3 signaling pathway in cancer and the development of specific inhibitors (Review). Oncol Lett. 2020;19(4):2585- 94.
77. Bharadwaj U, Kasembeli MM, Tweardy DJ. STAT3 Inhibitors in Cancer: A Comprehensive Update. STAT3 Inhibitors in Cancer. 2016;95-161.
78. Garg M, Shanmugam MK, Bhardwaj V, Goel A, Gupta R, Sharma A, et al. The pleiotropic role of transcription factor STAT3 in oncogenesis and its targeting through natural products for cancer prevention and therapy. Med Res Rev. 2020;1-46.
79. Fragoulis GE, McInnes IB, Siebert S. JAKinhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology. 2019;58(Suppl 1):i43-i54.
80. Buchert M, Burns CJ, Ernst M. Targeting JAK kinase in solid tumors: emerging opportunities and challenges. Oncogene. 2016;35(8):939-51.
81. Mascarenhas J, Hoffman R. Ruxolitinib: The First FDA Approved Therapy for the Treatment of Myelofibrosis. Clin Cancer Res. 2012;18(11):3008-14.
82. Dhillon S. Tofacitinib: A Review in Rheumatoid Arthritis. Drugs. 2017;77(18):1987-2001.
83. Maeda H. Japan’s Special Approval for Emergency System During the COVID-19 Pandemic. Clin Pharmacol Ther. 2021;10.1002/cpt.2310.
84. Horiguchi A, Asano T, Kuroda K, Sato A, Asakuma J, Ito K, et al. STAT3 inhibitor WP1066 as a novel therapeutic agent for renal cell carcinoma. Br J Cancer. 2010;102(11):1592-9.
85. McFarland BC, Ma JY, Langford CP, Gillespie GY, Yu H, Zheng Y, et al. Therapeutic potential of AZD1480 for the treatment of human glioblastoma. Mol Cancer Ther. 2011;10(12):2384-93.
86. Sen B, Saigal B, Parikh N, Gallick G, Johnson FM. Sustained Src Inhibition Results in Signal Transducer and Activator of Transcription 3 (STAT3) Activation and Cancer Cell Survival via Altered Janus-Activated Kinase- STAT3 Binding. Cancer Res. 2009;69(5):1958-65.
87. Santos FP, Kantarjian HM, Jain N, Manshouri T, Thomas DA, Garcia-Manero G, et al. Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood. 2010;115(6):1131- 6.
88. Quintás-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115(15):3109-17.
89. Kobayashi A, Tanizaki Y, Kimura A, Ishida Y, Nosaka M, Toujima S, et al. AG490, a Jak2 inhibitor, suppressed the progression of murine ovarian cancer. Eur J Pharmacol. 2015;766:63-75.
90. Fu LX, Lian QW, Pan JD, Xu ZL, Zhou TM, Ye B. JAK2 tyrosine kinase inhibitor AG490 suppresses cell growth and invasion of gallbladder cancer cells via inhibition of JAK2/STAT3 signaling. J Biol Regul Homeost Agents. 2017;31(1):51-8.
91. Derenzini E, Lemoine M, Buglio D, Katayama H, Ji Y, Davis RE, et al. The JAK inhibitor AZD1480 regulates proliferation and immunity in Hodgkin lymphoma. Blood Cancer J. 2011;1(12):e46.
92. Murakami T, Takigawa N, Ninomiya T, Ochi N, Yasugi M, Honda Y, et al. Effect of AZD1480 in an epidermal growth factor receptor-driven lung cancer model. Lung Cancer. 2014;83(1):30-6.
93. Plimack ER, Lorusso PM, McCoon P, Tang W, Krebs AD, Curt G, et al. AZD1480: A Phase I Study of a Novel JAK2 Inhibitor in Solid Tumors. Oncologist. 2013;18(7):819-20.
94. Gharibi T, Babaloo Z, Hosseini A, Abdollahpour- Alitappeh M, Hashemi V, Marofi F, et al. Targeting STAT3 in cancer and autoimmune diseases. Eur J Pharmacol. 2020;878:173107.
95. Johnson FM, Agrawal S, Burris H, Rosen L, Dhillon N, Hong D, et al. Phase 1 pharmacokinetic and druginteraction study of dasatinib in patients with advanced solid tumors. Cancer. 2010;116(6):1582-91.
96. Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and Efficacy of INCB018424, a JAK1 and JAK2 Inhibitor, in Myelofibrosis. N Engl J Med. 2010;363(12):1117-27.
97. Damsky W, Peterson D, Ramseier J, Al-Bawardy B, Chun H, Proctor D, et al. The emerging role of Janus kinase inhibitors in the treatment of autoimmune and inflammatory diseases. J Allergy Clin Immunol. 2021;147(3):814-26.
98. Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, Settleman J. Drug resistance via feedback activation of Stat3 in oncogeneaddicted cancer cells. Cancer Cell. 2014;26(2):207-21.
99. Tanizaki J, Okamoto I, Takezawa K, Sakai K, Azuma K, Kuwata K, et al. Combined effect of ALK and MEK inhibitors in EML4-ALK-positive non-small-cell lung cancer cells. Br J Cancer. 2012;106(4):763-7.
100. Witt K, Evans-Axelsson S, Lundqvist A, Johansson M, Bjartell A, Hellsten R. Inhibition of STAT3 augments antitumor efficacy of anti-CTLA-4 treatment against prostate cancer. Cancer Immunol Immunother. 2021;Mar 31.
101. Hayakawa T, Yaguchi T, Kawakami Y. Enhanced anti-tumor effects of the PD-1 blockade combined with a highly absorptive form of curcumin targeting STAT3. Cancer Sci. 2020;111(12):4326-35.
102. Chen TH, Chang PM, Yang MH. Novel immunemodulating drugs for advanced head and neck cancer. Head Neck. 2019;41 Suppl 1:46-56.
103. Mohrherr J, Uras IZ, Moll HP, Casanova E. STAT3: Versatile Functions in Non-Small Cell Lung Cancer. Cancers. 2020;12(5):1107.
104. Balic JJ, Albargy H, Luu K, Kirby FJ, Jayasekara WSN, Mansell F, et al. STAT3 serine phosphorylation is required for TLR4 metabolic reprogramming and IL-1β expression. Nat Commun. 2020;11(1):3816.
105. Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports RAS-dependent oncogenic transformation. Science. 2009;324(5935):1713-6.
106. Reich NC. STATs get their move on. JAKSTAT. 2013;2(4):e27080.
107. Brachet-Botineau M, Polomski M, Neubauer HA, Juen L, Hédou D, Viaud-Massuard MC, et al. Pharmacological Inhibition of Oncogenic STAT3 and STAT5 Signaling in Hematopoietic Cancers. Cancers. 2020;12(1):240.
108. Koseki T, Suehiro N, Masuda Y, Miyoshi N, Muraoka D, Ogo N, et al. Discovery of a New STAT3 Inhibitor Acting on the Linker Domain. Biol Pharm Bull. 2019;42(5):792- 800.
109. Ashizawa T, Akiyama Y, Miyata H, Iizuka A, Komiyama M, Kume A, et al. Effect of the STAT3 inhibitor STX-0119 on the proliferation of a temozolomide-resistant glioblastoma cell line. Int J Oncol. 2014;45(1):411-8.
110. Li W, Yang H, Li X, Han L, Xu N, Shi A. Signaling pathway inhibitors target breast cancer stem cells in triplenegative breast cancer. Oncol Rep. 2019;41(1):437-46.
111. Wu DM, Zheng ZH, Zhang YB, Fan SH, Zhang ZF, Wang YJ, et al. Down-regulated lncRNA DLX6-AS1 inhibits tumorigenesis through STAT3 signaling pathway by suppressing CADM1 promoter methylation in liver cancer stem cells. J Exp Clin Cancer Res. 2019;38(1):237.
112. Liang M, Zhan F, Zhao J, Li Q, Wuyang J, Mu G, et al. CPA-7 influences immune profile and elicits anti-prostate cancer effects by inhibiting activated STAT3. BMC Cancer. 2016;16:504.
113. Shastri A, Choudhary G, Teixeira M, Gordon- Mitchell S, Ramachandra N, Bernard L, et al. Antisense STAT3 inhibitor decreases viability of myelodysplastic and leukemic stem cells. J Clin Invest. 2018;128(12):5479-88.
114. Li X, Wei Y, Wei X. Napabucasin, a novel inhibitor of STAT3, inhibits growth and synergises with doxorubicin in diffuse large B-cell lymphoma. Cancer Lett. 2020;491:146- 61.