Abstract
ZBP1 has aroused a wide interest since it was identified as the first cytosolic dsDNA sensor ahead of the finding of cGAS in 2013. However, the investigations on ZBP1 declined when researchers found the binding of ZBP1 with dsDNA is not absolutely required for the activation of innate response. Recently, the significance of ZBP1 was reemphasized as a key regulator of innate immunity. Here we reviewed the emergence of evidence on how ZBP1 promotes cell death and inflammation upon physiological and pathological challenges.
Keywords
ZBP1, Cell death, Inflammation, Cytosolic NA sensor
Abbreviation
ZBP1: Z-DNA Binding Protein 1; dsDNA: Double Strand Deoxyribonucleic acid; cGAS: Cyclic GMP-AMP Synthase; ADAR1: Adenosine Deaminases Acting on RNA; IFN: Interferon; HSV: Herpes Simplex Virus; DAI: DNA-dependent activator of IFN (interferon)- regulatory factor; AIM2: Absent In Melanoma 2; RHIMs: Receptor-interacting protein (RIP) Homotypic Interaction motifs; TNF: Tumor Necrosis Factor; MCMV: Murine Cytomegalovirus Virus; MEFs: Mouse Embryonic Fibroblasts; DVGs: Defective Viral Genomes; IAV: Influenza A Virus; ERVs: Endogenous Retroviruses; HSV1: Herpes Simplex Virus 1; dsRNA: Double strand Ribonucleic Acid
Importance
TNFs promote programmed cell death (PCD), including apoptosis and necroptosis under certain circumstance [1-4]. Evidence showed that TNF-induced cell death is the pathology of certain inflammatory diseases [4]. However, some inflammation caused by cell death are TNF-independent. Recently, several groups reported that ZBP1, once thought to be a DNA sensor, triggers PCD and inflammation upon virus infection or endogenous retrovirus activation [5-8]. Understanding the pathologic role of ZBP1 may contribute to the treatment of incurable inflammation in clinic.
The Tale of ZBP1
ZBP1 was named DLM-1 when it was first cloned as a tumorassociated protein in 1999 [9]. ZBP1 was highly expressed in the peritoneal lining tissue of ovarian ascites tumors and believed to mediate the process of host responses to tumorigenesis [9]. Two years later, Rich group reported that the N-terminus of DLM-1 contains two Zα high affinity binding domains, which is highly conserved with that of RNA editing enzyme ADAR1 by crystal structure comparison [10]. Crystal structure of ZαDLM-1 binding with left-handed Z-DNA gives DLM-1 another name ZBP1 (Z-DNA Binding Protein 1) and provokes intensive investigations on dsDNA-sensing function of ZBP1 physiologically and pathologically. ZBP1 was called DAI and once considered as the first cytosolic DNA sensor in 2007 when cGAS had not been identified yet [11]. Taniguchi lab and others reported the binding of ZBP1 with cytosolic dsDNA, which activates innate immune response through type-I interferon and NF-κB pathways. Takaoka further showed the requirement of DAI for the production of type I interferon in L929 cells upon HSV infection, however, in most of the viral infected or plasmid transfected cells, the capacity of DAI to combine with B-DNA and plasmid DNA is indispensable for cells to activate innate immunity because of redundant cytosolic B-DNA sensors, such as cGAS and AIM2 etc. [12-14]. Therefore, in this review, we call it ZBP1 hereinafter.
In addition to two Zα domains at the N-terminal for nucleic acid (NA) binding, its c-terminus domain (CTD) recruits IRF3 and TBK1 to activate the type I IFN pathway (Figure 1) [11]. There are two RHIMs located in the middle of ZBP1, which were first identified as sites for ZBP1 recruitment of RIP1 and RIP3 to activate NF-κb in 2008 (Figure 1) [15]. RIP proteins, a family of receptor-interacting serine/threonine-protein kinases, bind with ZBP1 or with each other by their RHIM domains. RIP1/RIP3 play an essential role in programmed cell death, especially in TNF -driven necroptosis [16], which has been well documented over the past two decades [17-19]. Thus, activated ZBP1 recruits RIP3 for necroptosis; or RIP3/RIP1 and Caspase 8 to mediate apoptosis [20]. So far, ZBP1 is the only nucleic acid sensor that contains RHIM domains [18,21] to directly trigger cell death under extracellular or intracellular stimuli.
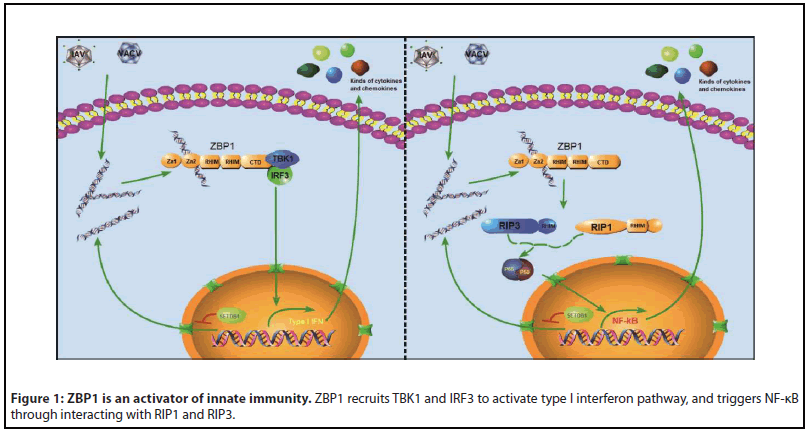
ZBP1 Activation under Extracellular Stimulation
As a cytosolic NA sensor, the function of ZBP1 was discovered largely during viral infection. ZBP1 is an activator of innate immunity in response to infection of DNA or RNA viruses, such as vaccinia, Human Cytomegalovirus, IAV etc. [20,22,23]. IAVinfected MEFs (mouse embryonic fibroblasts) or BMDMs (bone marrow derived macrophages) undergo ZBP1-dependent necroptosis, apoptosis and pyroptosis, which is the strategy that the host defense controls the dangerous infections by eliminating the places for viral production [20,24]. Therefore, under the circumstances of IAV infection, mice that are deficient of ZBP1 or with a defect in ZBP1-triggered cell death suffer from severer inflammation and die in higher ratio of lethality [20,25]. ZBP1 was upregulated by viral infection, but it is not sufficient for the induction of ZBP1-triggered cell death. For example, M45 was a Murine Cytomegalovirus Vironassociated protein, which contains a RHIM domain that it can inhibit ZBP1-triggered necroptosis. In primary MEFs infected with MCMV-M45 RHIM mutation, ZBP1 triggered necroptosis was blocked when the Zα domain of ZBP1 was absent [26], suggesting the Zα domain is also required for ZBP1-driven cell death in MCMV-infected cells. The answer of ZBP1 activation upon viral infection is revealed last year. A study reported that DVGs was released from rapid replication of IAVs genome, which formed Z-RNAs that combined to the Zα domain of ZBP1 [5]. Then the NA bound ZBP1 recruited RIP3 via their RHIM domains for cell death. It should be noted that cell death is not the only destination upon ZBP1 activation by viral infection. Metabolic reprogram by ZBP1-IRF1-IRG1 axis was reported in ZIKA-viral infected neurons to eliminate virus and keep neurons alive [27]. In addition to response to viral infection, numerous studies reported that ZBP1 also plays a role against the infections of bacteria and fungi with an unclear mechanism [28,29].
Endogenous Ligand Triggered ZBP1-death Pathway
RIP1 promotes TNF-triggered necroptosis in a kinase activitydependent manner [30]. Unexpectedly, four groups found that RIP1 blocks early embryonic lethality and inflammation, and it maintains the homeostasis of skin and intestine against inflammation and cell death, which implies that RIP1 plays a negative role in TNF-independent necroptosis [31-34]. The following work in 2016 by Lin et al. and Newton et al. reported that the TNF-independent necroptosis which can be halted by the RHIM of RIP1 is ZBP1-dependent [35,36]. Interestingly, the Zα2- mutated ZBP1 mice are as resistant as ZBP1 deleted mice on skin inflammation caused by RIP1-RHIM mutation [8], which suggested that there should be some endogenous ligands that physically or pathologically ignited ZBP1 via Zα domain in non-viral infected cells. In 2020, Wang et al. found the key in intestinal epithelial cells with reactive endogenous retroviruses (ERVs) by Setdb1 deletion [7]. SETDB1, a histone H3 lysine 9 (H3K9) trimethylation methyltransferase, maintains the H3K9me3 modification along genome to silence the TEs ( transposon elements) against genome instability [37]. ERVs (endogenous retroviruses) are one of the TEs and occupy nearly 10% of the genome [7]. Setdb1 deficiency initiates ZBP1- triggered necroptosis in intestinal stem cells. The death could be blocked by Zα mutation of ZBP1, indicating the binding of death signal to Zα domain of ZBP1. It turns out the dsRNAs transcribed from deactivated ERVs are the ligand of ZBP1 for epithelial cell death. Although it is yet unclear whether the ERVs-dsRNAs form Z conformation or not, the conclusion that ERVs-dsRNAs are the endogenous ligand of ZBP1 is supported by another group. They reported that death of keratinocytes and skin inflammation by RIP1-RHIM mutation were triggered by the interaction between trace ERVs-dsRNA and ZBP1 [8].
Negative Regulation of ZBP1-dependent Cell Death
From the perspective of infection by exogenous ligand, to counter the ZBP1-RIP3 mediated cell death, various strategies have been adopted by virus. ICP6, a RHIM-containing protein from HSV1, promotes RIP3-mediated necroptosis in murine cells but inhibits necroptosis in human cells [38,39]. Vira, another viral-RHIM protein transcribed from a viral gene M45 of murine cytomegalovirus (MCMV) competes with RIP3 for the binding to ZBP1 and suppresses ZBP1-RIP3-mediated necroptosis (Figure 2) [26]. Different from HSV1 and MCMV, vaccinia virus (VACV) blocks ZBP1-triggered necroptosis by a strategy to compete for viral nucleic acid with ZBP1 by a Zα domain and a dsRNA binding domain-containing protein E3L (Figure 2) [40]. Both of nucleic acid binding domains collaborate to inhibit ZBP1-triggered necroptosis, whose mutations result in cell death by VACV infection [41]. It is such a marvelous and delicate revolution of competition between the host and pathogen that are superbly present in the ways of viral regulation on ZBP1-RIP3 mediated cell death. To the respect of activation by endogenous ligand, RIP1 inhibits the ZBP1-RIP3 necroptosis pathway by its RHIM domain [36]. It has been reported that ZBP1-triggered necroptosis can be induced by IFNs in either RIP1, Caspase 8 or FADD deficient L929 cells (Figure 2) [42]. In the FADD/Caspase 8 knockout intestinal epithelial cells, epithelial homeostasis broke down with intestinal inflammation which are related with ZBP1-triggered cell death [43]. It implies that RIP1/FADD/ CASPASE 8 may function as a negative regulator of ZBP1-RIP3 cell death. However, the detail of the protective mechanism against cell death remains to be explored. Recently, several individual studies revealed that RNA deaminase ADAR1 edits endogenous Z-form RNA [44-46], suggesting ADAR1 might be a negative regulator by degradation the ligand of ZBP1.
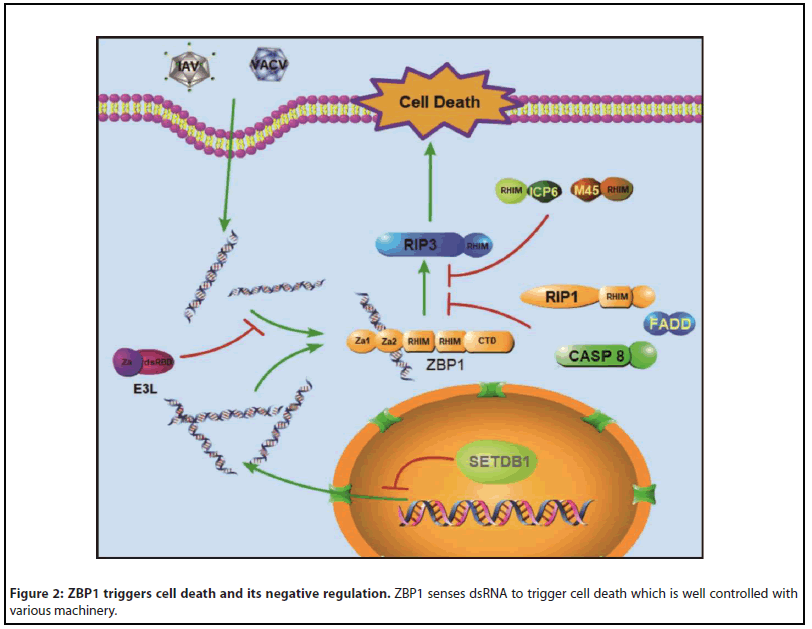
ZBP1 and Multiple Death Types
PAN-optosis refers to the collective activation of apoptosis, necroptosis and inflammasome/pyroptosis, which occurs upon IAV infections and can be mediated by ZBP1 [47]. In the MEFs infected by IAV, apoptosis and necroptosis occur in different cells. Pyroptosis takes place in the infected BMDMs, which was blocked by ZBP1 deletion or NLRP3 deletion [24]. But the connection between ZBP1 and NLRP3 inflammasome is unknown yet. ZBP1 could assemble PAN-optosome including RIP3, RIP1, FADD and Caspase8 to trigger PANoptosis [6]. Caspase 6 was reported to regulate the assembly of PAN-optosome. Although IAVs infections lead to ZBP1- induced PAN-optosis, whether the ERVs-dsRNAs share the same story is unclear. Deepening the study of ZBP1-triggered endogenous cell death in uninfected cell will benefit a lot for the understanding and clinic treatment of related autoinflammation disease [48]. Different types of cell death have different physiological influences. In IAVs infection, ZBP1- triggered apoptosis is helpful for viral clearance and protection of the host. In contrast, ZBP1-initiated necroptosis provokes the acute inflammation and lethality [5]. Understanding the key steps that determine the cell fate under ZBP1-triggered PAN-optosome would benefit infection control as well as the compromise of the hyperactivation of innate immunity.
Prospect
Lacking the binding pockets in structure, ZBP1 is difficult to be suppressed by small molecular compounds. The strategy of manipulating the ZBP1 should be more focused on the ligand that activates ZBP1 or proteins that regulates the activation of ZBP1 positively or negatively. Screening the compounds that targeting the dsRNA degradation system may have good prospects in inflammatory disease and tumor immunity.
References
2. Newton K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 2015;25(6):347-53.
3. Tummers B, Green DR. Caspase-8: regulating life and death. Immunol Rev. 2017;277(1):76-89.
4. Zhou W, Yuan J. Necroptosis in health and diseases. Semin Cell Dev Biol. 2014;35:14-23.
5. Zhang T, Yin C, Boyd DF, Quarato G, Ingram JP, Shubina M, et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell. 2020;180(6):1115-29 e13.
6. Zheng M, Karki R, Vogel P, Kanneganti TD. Caspase-6 Is a Key Regulator of Innate Immunity, Inflammasome Activation, and Host Defense. Cell. 2020;181(3):674-87.e13.
7. Wang R, Li H, Wu J, Cai ZY, Li B, Ni H, et al. Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature. 2020;580(7803):386-90.
8. Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature. 2020;580(7803):391-5.
9. Fu Y, Comella N, Tognazzi K, Brown LF, Dvorak HF, Kocher O. Cloning of DLM-1, a novel gene that is up-regulated in activated macrophages, using RNA differential display. Gene. 1999;240(1):157-63.
10. Schwartz T, Behlke J, Lowenhaupt K, Heinemann U, Rich A. Structure of the DLM-1-Z-DNA complex reveals a conserved family of Z-DNA-binding proteins. Nat Struct Biol. 2001;8(9):761-5.
11. Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448(7152):501-5.
12. Lippmann J, Rothenburg S, Deigendesch N, Eitel J, Meixenberger K, van Laak V, et al. IFNbeta responses induced by intracellular bacteria or cytosolic DNA in different human cells do not require ZBP1 (DLM- 1/DAI). Cell Microbiol. 2008;10(12):2579-88.
13. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, et al. cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498(7454):380-4.
14. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458(7237):514-8.
15. Kaiser WJ, Upton JW, Mocarski ES. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J Immunol. 2008;181(9):6427-34.
16. Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. 2009;138(2):229-32.
17. Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332-6.
18. He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100-11.
19. Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135(7):1311-23.
20. Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3- Dependent Cell Death. Cell Host Microbe. 2016;20(5):674-81.
21. Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 2009;10(8):916-22.
22. DeFilippis VR, Alvarado D, Sali T, Rothenburg S, Fruh K. Human cytomegalovirus induces the interferon response via the DNA sensor ZBP1. J Virol. 2010;84(1):585-98.
23. Kim YG, Muralinath M, Brandt T, Pearcy M, Hauns K, Lowenhaupt K, et al. A role for Z-DNA binding in vaccinia virus pathogenesis. Proc Natl Acad Sci U S A. 2003;100(12):6974-9.
24. Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1(2).
25. Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH, 3rd, Ingram JP, et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe. 2016;20(1):13-24.
26. Pham CL, Shanmugam N, Strange M, O'Carroll A, Brown JW, Sierecki E, et al. Viral M45 and necroptosis-associated proteins form heteromeric amyloid assemblies. EMBO Rep. 2019;20(2).
27. Daniels BP, Kofman SB, Smith JR, Norris GT, Snyder AG, Kolb JP, et al. The Nucleotide Sensor ZBP1 and Kinase RIPK3 Induce the Enzyme IRG1 to Promote an Antiviral Metabolic State in Neurons. Immunity. 2019;50(1):64-76 e4.
28. Muendlein HI, Connolly WM, Magri Z, Smirnova I, Ilyukha V, Gautam A, et al. ZBP1 promotes LPS-induced cell death and IL-1beta release via RHIM-mediated interactions with RIPK1. Nat Commun. 2021;12(1):86.
29. Cervantes PW, Martorelli Di Genova B, Erazo Flores BJ, Knoll LJ. RIPK3 Facilitates Host Resistance to Oral Toxoplasma gondii Infection. Infect Immun. 2021;89(5).
30. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11(10):700-14.
31. Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157(5):1189-202.
32. Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, Wachsmuth L, et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513(7516):90-4.
33. Rickard JA, O'Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell. 2014;157(5):1175-88.
34. Takahashi N, Vereecke L, Bertrand MJ, Duprez L, Berger SB, Divert T, et al. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature. 2014;513(7516):95-9.
35. Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, Pham VC, et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature. 2016;540(7631):129-33.
36. Lin J, Kumari S, Kim C, Van TM, Wachsmuth L, Polykratis A, et al. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature. 2016;540(7631):124-8.
37. Cuellar TL, Herzner AM, Zhang X, Goyal Y, Watanabe C, Friedman BA, et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J Cell Biol. 2017;216(11):3535-49.
38. Wang X, Li Y, Liu S, Yu X, Li L, Shi C, et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci U S A. 2014;111(43):15438-43.
39. Ali M, Roback L, Mocarski ES. Herpes simplex virus 1 ICP6 impedes TNF receptor 1-induced necrosome assembly during compartmentalization to detergent-resistant membrane vesicles. J Biol Chem. 2019;294(3):991-1004.
40. Koehler H, Cotsmire S, Langland J, Kibler KV, Kalman D, Upton JW, et al. Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc Natl Acad Sci U S A. 2017;114(43):11506-11.
41. Koehler H, Cotsmire S, Zhang T, Balachandran S, Upton JW, Langland J, et al. Vaccinia virus E3 prevents sensing of Z-RNA to block ZBP1-dependent necroptosis. Cell Host Microbe. 2021.
42. Yang D, Liang Y, Zhao S, Ding Y, Zhuang Q, Shi Q, et al. ZBP1 mediates interferon-induced necroptosis. Cell Mol Immunol. 2020;17(4):356-68.
43. Schwarzer R, Jiao H, Wachsmuth L, Tresch A, Pasparakis M. FADD and Caspase-8 Regulate Gut Homeostasis and Inflammation by Controlling MLKL- and GSDMD-Mediated Death of Intestinal Epithelial Cells. Immunity. 2020;52(6):978-93.e6.
44. Tang Q, Rigby RE, Young GR, Hvidt AK, Davis T, Tan TK, et al. Adenosine-to-inosine editing of endogenous Z-form RNA by the deaminase ADAR1 prevents spontaneous MAVS-dependent type I interferon responses. Immunity. 2021;54(9):1961-75.e5.
45. Nakahama T, Kato Y, Shibuya T, Inoue M, Kim JI, Vongpipatana T, et al. Mutations in the adenosine deaminase ADAR1 that prevent endogenous Z-RNA binding induce Aicardi-Goutières-syndrome-like encephalopathy. Immunity. 2021;54(9):1976-88.e7.
46. Maurano M, Snyder JM, Connelly C, Henao-Mejia J, Sidrauski C, Stetson DB. Protein kinase R and the integrated stress response drive immunopathology caused by mutations in the RNA deaminase ADAR1. Immunity. 2021;54(9):1948-60.e5.
47. Malireddi RKS, Kesavardhana S, Kanneganti TD. ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis). Front Cell Infect Microbiol. 2019;9:406.
48. Samir P, Malireddi RKS, Kanneganti TD. The PANoptosome: A Deadly Protein Complex Driving Pyroptosis, Apoptosis, and Necroptosis (PANoptosis). Front Cell Infect Microbiol. 2020;10:238.