Abstract
Traditional catalytic theory has predominantly centered on static chemical bond processes, specifically the weakening, breaking, strengthening, and formation of chemical bonds – representing relatively fixed patterns of transformation. Whereas the dynamic regulatory mechanisms of weak interactions have been relatively understudied. This paradigm shift emerges from recognizing how precisely engineered 3D spatial arrangements govern catalytic efficiency: directional hydrogen bonds, size-matched hydrophobic cavities, and π-π stacking at optimal distances collectively create confined microreactors that steer reaction pathways. Through systematic analysis of hydrogen bond strength gradation, synergistic multicomponent coupling and cross-scale systems (such as the catalyst phosphonium chalcogenide 9 (PCH9), hydrogen-bonded organic frameworks) supported by theoretical and experimental evidence, we propose: (1) Strong hydrogen bonds can rigidify molecular networks to selectively stabilize intermediates, whereas weak interactions dynamically optimize interfacial microenvironments; (2) Cooperative weak interactions can activate inert substrates and enable efficient charge/proton transfer; (3) Weak-interaction networks provide a scalable design framework for selectivity across molecular to mesoscale systems. This paradigm establishes a universal mechanistic framework for catalysis beyond traditional static bond models. In future work, operando spectroscopy should be employed to quantify transient weak interactions lifetimes and facilitate scalable applications in sustainable fine chemical synthesis.
Keywords
Weak interactions, Dynamic regulatory mechanisms, Hydrogen bond, Interfacial microenvironment, Catalysis, Synergistic multicomponent coupling, Selectivity
Commentary
Traditional chemical research has predominantly focused on static chemical bond transfer processes. However, the inherent complexity of catalytic systems is shifting the research paradigm from static descriptions to mechanistic dynamics analysis. Contemporary studies reveal that catalytic reactions extend far beyond simple bond activation: they are intrinsically linked to the dynamic evolution of molecular configurations, synergistic regulation of multicomponent coupling effects, conformational reorganization during bimolecular adsorption, and challenges like pressure regime disparity, material scaling challenges, and multiscale complexity issues under specific conditions. Notably, transient transition states and intermediates, which are key nodes connecting reactants and products in in situ reaction processes, are often overlooked in traditional catalytic mechanism studies, particularly in the insufficient analysis of weak interaction-driven dynamic structural evolution during transition state exploration. This inadequacy arises from an overemphasis on static chemical bond transformations, which neglects the multiscale regulatory roles of weak interactions-including molecular conformational reorganization, interfacial microenvironment modulation, and transition state stabilization. Consequently, mechanistic analyses often fail to link interaction dynamics to experimental outcomes. Deeper exploration of such correlations is therefore imperative.
Although weak interactions are characterized by short lifetimes and low energetic contributions, precise control over intermolecular directional recognition and dynamic matching offers significant insights into catalysis. Such control could enable the construction of efficient energy transfer pathways and conformational guidance within reaction pathways. A thorough investigation and systematic elucidation of the mechanisms underlying these dynamic processes will not only deepen our fundamental understanding of catalysis but also provide critical theoretical foundations for the rational design of industrial catalytic systems.
Catalytic reactions essentially involve dynamically coordinated networks of intermolecular interactions that precisely modulate energy transfer and conformational evolution. A critical research gap that demands attention is the structure-performance correlation between weak interactions and catalytic function. The energy network influenced by these weak interactions encompasses van der Waals forces (dispersion forces (Figure 1A), induction forces (Figure 1B), orientation forces (Figure 1C), hydrogen bonding (Figure 1D), π-π stacking, electrostatic interactions, and hydrophobic effects. Their picosecond-scale, time-resolved dynamic response characteristics can directionally lock transition states and optimize mass transfer pathways, thereby offering novel strategies to address long-standing selectivity challenges.
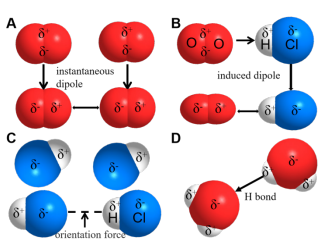
Figure 1. Schematic diagram of van der Waals forces and hydrogen bonds (A) Dispersion forces. (B) Induction forces. (C) Orientation forces. (D) Hydrogen bonding.
Among these interactions, hydrogen bonding serves as a core regulatory element due to its directionality and dynamic adaptability, making it an ideal probe for deciphering the mechanisms of weak interactions. Classified by bond strength, hydrogen bonds range from strong to weak, and their synergistic interplay forms a central hub for precise pathway regulation in catalysis.
Strong hydrogen bonds can rigidify molecular networks to govern macroscopic processes. As demonstrated by Yang et al. [1], *OH groups were rationally engineered near oxygen reduction reaction (ORR) active sites as hydrogen bond acceptors. The electronic structure of these groups—particularly the lone pair electron density and electronegativity of oxygen atoms—determines their strength as hydrogen bond acceptors. By leveraging the precise spatial match between the extended conformation of *OOH and the effective range of hydrogen bonding (Figure 2A), the distance between the implanted *OH and the *OOH intermediate was confined within the hydrogen bonding range (<2 Å), while exceeding this range for the *OH intermediate. This design achieved selective stabilization of the *OOH intermediate exclusively via hydrogen bonding, thereby enabling targeted binding of the ORR intermediate *OOH (theoretical).
On palladium catalyst surfaces, cysteamine ligands form a rigid N···H–N hydrogen bond network via intramolecular hydrogen bonding. The unique steric hindrance induced by the hydrogen bond network hinders alkene adsorption while enabling alkyne hydrogenation to follow the anti-Markovnikov’s rule, achieving high semi hydrogenation selectivity (>99% alkene yield) [2] (experimental). The regulation of proton-coupled electron transfer (PCET) kinetics by interfacial hydrogen-bond networks in protic ionic liquids [3] and the acceleration of proton transfer in hydrogen evolution reaction (HER) via crown ether–water networks further underscore the universal regulatory mechanism of strong hydrogen bonding [4] (experimental).
Compared to the rigid control exerted by strong hydrogen bonds, weak hydrogen bonds exhibit superior adaptability to microenvironmental changes. At the water/b-TiO2 (210) interface system (Figure 2B) [5], weakened hydrogen bonding drives the selective generation of H2O2 through a triple mechanism: (i) extending the hydrogen bond distance between *OH and water to 1.54 Å. This effect stems from interfacial water's electronic structures. Ti³+ defects or specific TiO2 facets alter surface oxygen's electronic/geometric properties, reducing hydrogen-bonding capability with water hydrogens. Such surface electronic modulation enables the "weakened hydrogen bond connectivity" strategy for selective H2O2 production; (ii) forming a herringbone-like surface structure that creates a low water density cavity to hinder deprotonation; (iii) enhancing the adsorption energy of *OH to lower the coupling barrier (theoretical). This advantage of flexible regulation is further demonstrated in cinchoninium catalysis, where the electrophilic enal is anchored via C–H···O ion-pair interactions, and a peripheral non-classical hydrogen bond network (seven weak interactions) confines the nucleophile [6] (Figure 2C). This synergistic effect collectively lowers the Gibbs free energy of the transition state, thereby overcoming the selectivity issue in imine polarity reversal (theoretical).
The spatiotemporal dynamics of weak interactions is equally crucial at heterogeneous interfaces and throughout the entire reaction process. The interfacial O–H···O weak hydrogen bonds in the BiOBr/NiFe-LDH heterojunction (Figure 2D) [7] not only promote charge transfer but also ensure stability over 50 cycles of stability (experimental). This transition point from weak to strong interactions constitutes a key nexus for boosting both the activity and stability of the catalytic system, fundamentally determining the efficient execution of the catalytic reaction.
In the catalytic process, weak interactions play key regulatory roles at different stages. Before adsorption, via the solvation effect, weak electrostatic interactions pre-organize reactant configurations, reducing the adsorption entropy barrier and prompting molecules to contact the active sites in the optimal orientation. During the transition state, weak hydrogen bonds dynamically stabilize intermediates [8]. As the reaction proceeds, the insufficiency of single weak interactions necessitates synergistic strategies across scales, thereby enhancing catalyst durability.
In situ characterization can capture the structural evolution of catalysts, such as the formation of Co-O sites in Co-based photocatalysis, and track weak interactions [9] (experimental). Operando Raman and other techniques can reveal dynamic bond formation and quantify transient weak interactions, helping understand their roles in activity/selectivity, which is crucial for advancing catalytic mechanisms.
Beyond regulating catalytic steps, hydrogen bonds play a crucial role in directing the synthesis and assembly of catalytic materials themselves. For instance, in the formation of Ni-1,3, 5-benzenetricarboxylate (Ni-BTC) nanosheets, the hydrogen bonding between ethanol and BTC ligands inhibits BTC deprotonation, directing nanosheet growth. Simultaneously, hydrogen bonds between hydroxyl groups on the Ti mesh's TiO2 layer and BTC carboxyl groups dehydrate under solvothermal conditions, converting to coordination bonds that induce in situ nucleation and drive nanosheet array self-assembly [10] (experimental).
Similarly, in the CoFe-5,5′,5′’-(1,3,5-triazine-2,4,6-triyl) tris(azanediyl)triisophthalic acid (CoFe-TDPAT) MOF system, hydrogen bonding drives catalytic performance through a triple synergy mechanism [11]: The secondary amine groups of H6TDPAT form intermolecular hydrogen bonds (FTIR red shift to 3414.5 cm-¹), enhancing framework stability (ensuring 100h alkaline environment durability); the hydrogen bonding between methanol solvent and TDPAT slows metal coordination rates (compared to DMF); the hydrogen bond network induces superhydrophilic interfaces that suppress O2/H2 bubble adhesion, accelerating mass transfer efficiency under high current densities of 300 mA cm-2 (experimental). These examples highlight how hydrogen bonding orchestrates both the *construction* of catalytic architectures and their subsequent function.
Beyond hydrogen bonding, other weak interactions such as hydrophobic effects play indispensable roles in catalytic selectivity modulation. Hydrophobic interactions demonstrate equally critical value [12]: The hydrophobic cavity of β-cyclodextrin achieves dynamic self-assembly through specific recognition of hydrophobic groups (such as adamantyl) in di (1-adamantyl) benzylphosphine (DABP) (Figure 2E). This supramolecular strategy demonstrates how weak interactions can fine-tune catalyst performance, significantly enhancing the selectivity for linear aldehyde formation in the corresponding catalytic system (experimental).
However, when confronted with the challenge of activating inert substrates, a single weak interaction often proves insufficient due to its limited strength. To overcome this limitation, research has revealed that designing synergistic systems involving multiple weak interactions is a key strategy. As exemplified by the catalyst PCH9 (Figure 2F) developed by Zhao et al., which leverages cooperative Se···O and H···O interactions for ester activation [13], PCH9 activates the electrophilic site via Se···O interaction (lactone carbonyl) while its sulfonamide moiety forms an H···O hydrogen bond with the alcohol initiator, enhancing the latter’s nucleophilicity (experimental). This dual activation enables efficient ring-opening polymerization of ε-caprolactone at room temperature. Concurrently, chalcogen-bonding catalysis, as a strategy utilizing weak interactions, has also been successfully introduced into the field of carbene chemistry.
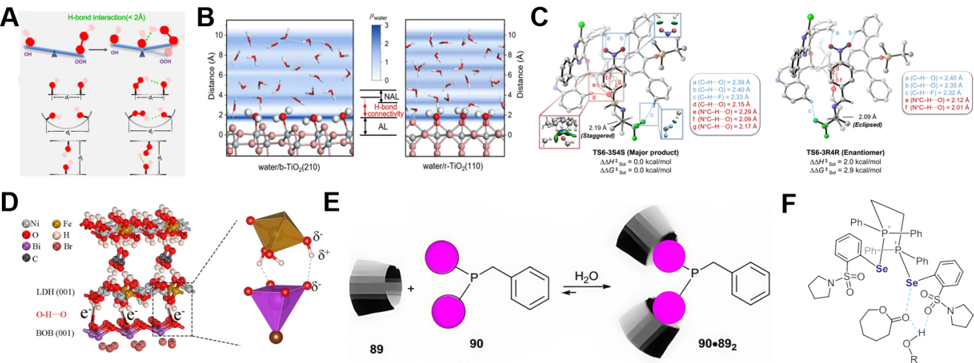
Figure 2. Schematic diagram of weak interactions and cooperative regulation mechanisms. (A) The strong hydrogen bonds stabilizing the intermediate. (B) The weak hydrogen bonds regulating the microenvironment at the liquid-solid interface. (C) Synergistic catalysis via C−H···O ion pairs and peripheral non-classical hydrogen bonds. (D) The weak hydrogen bonds facilitating charge transfer. (E) The hydrophobic cavity of dimethylated-β-CD (β-DIME) (89) interacts with the adamantyl group of the DABP (90) ligand through hydrophobic interactions. (F) The synergistic effect of Se···O chalcogenide bonds and N-H···O hydrogen bonds. (A) Reprinted with permission from Ref. [1], Copyright 2023, ChemSusChem. (B) Reprinted with permission from Ref. [5], Copyright 2024, Journal of the American Chemical Society. (C) Reprinted with permission from Ref [6], Copyright 2025, CCS Chemistry. (D) Reprinted with permission from Ref. [7], Copyright 2024, Angewandte Chemie International Edition. (E) Reprinted with permission from Ref. [12], Copyright 2014, Chemical Society Reviews. (F) Reprinted with permission from Ref. [13], Copyright 2024, Journal of the American Chemical Society.
This synergistic design concept extends further into the realm of mesoscale materials: In hydrogen-bonded organic frameworks (HOFs) [14], hydrogen bonds can serve to fix the pyrene rings, forming a stable two-dimensional layered framework. This hydrogen-bonded network provides the structural foundation for extending π-π stacking. Long-range π-π stacking in HOFs boosts electron delocalization and density, reducing the exciton binding energy (68.2 to 58.9 meV) to suppress electron-hole recombination and enhance charge separation/transport efficiency. Simultaneously, it increases electron density of loaded Pd NPs (evidenced by negative binding energy shift), enhancing their redox activity (experimental). The synergistic interplay between these two mechanisms could optimize the migration efficiency of photogenerated charge carriers, leading to an order-of-magnitude enhancement in catalytic activity.
In single nickel atom catalysis for CO2 reduction, electrostatic effects and hydrogen bonding synergize to boost activity (experimental) [15]. Surface charge (electrostatic force) endows sites with charge capacity; 0N and 1N sites accumulates 2e-, lowering barriers for *COOH/*CO formation (theoretical). This electrostatic enrichment force stabilizes polar intermediates like *COO. Hydrogen bonds, via water networks, anchor these intermediates, aiding H transfer from water (per docs). Electrostatically driven proximity of polar water to charged sites enhances H-bond dynamics, reinforcing intermediate stabilization.
This synergistic strategy of weak interactions spans multiple scales from the molecular-scale PCH9 catalyst (chalcogen/hydrogen bonding synergy), to mesoscale HOFs (hydrogen bonding/π-π stacking synergy), and up to system-level micellar nanoreactors (electrostatic/hydrophobic synergy) etc. Weak interactions and their synergistic strategies have established a comprehensive validation framework within catalysis [16,17].
Given the dynamic nature (picosecond scale) and low energetic contributions (typically far below those of covalent/ionic bonds) of weak interactions, precise elucidation of their structural evolution, energetic contributions, and synergistic mechanisms throughout catalytic processes (including adsorption, transition state stabilization, and product desorption stages) is crucial for validating the aforementioned regulatory strategies. This necessitates a synergistic approach combining advanced theoretical simulations with multiscale operando/in situ characterization techniques. For hydrogen bonds, density functional theory (DFT) calculations and ab initio molecular dynamics (AIMD) simulations have been used to analyze bond strengths, geometries, and dynamic behaviors at interfaces such as water/b-TiO2(210) [5]; operando spectroscopy and in situ measurements help quantify transient hydrogen bond lifetimes during catalytic processes. Hydrophobic interactions, as observed in β-cyclodextrin systems, are characterized using NMR spectroscopy to confirm host-guest recognition and surface tension measurements to assess molecular aggregation [12]. For synergistic weak interactions like Se···O and H···O in PCH9 catalyst, X-ray crystallography and DFT calculations reveal the cooperative bonding modes. Additionally, in hydrogen-bonded organic frameworks (HOFs), spectroscopy techniques and electron microscopy help unravel the interplay between hydrogen bonds and π-π stacking [14]. These characterization methods collectively provide insights into how weak interactions modulate catalytic activity, selectivity, and stability.
Conclusions
Traditional catalytic theory, constrained by static chemical bond models, has overlooked the dynamic regulatory role of weak interactions in governing catalytic efficiency. Through systematic analysis of hydrogen bond gradation, synergistic coupling, and cross-scale systems, this study demonstrates that:
- Strong hydrogen bonds rigidify molecular networks to selectively stabilize intermediates, while weak interactions dynamically optimize interfacial microenvironments;
- Cooperative weak interactions activate inert substrates and enable efficient charge/proton transfer;
- Weak-interaction networks provide a scalable design paradigm for selectivity control from molecular to mesoscale systems.
These findings establish a dynamic mechanistic framework beyond static bond models, bridging structural evolution with catalytic performance. Further validation through operando spectroscopy and multi-scale simulations is essential to address transient interaction dynamics and industrial scalability challenges.
Perspectives
In the future, as the fine chemical industry continues to increase demands for product performance and greener processes, the dynamic modulation capabilities of weak interactions will become increasingly prominent. They could serve not only as a critical nexus bridging traditional catalytic systems with novel high-efficiency catalytic systems, but also as a core driving force propelling the industry toward high-end development. During the ongoing transition toward “weak processes” in the fine chemical domain, the precise modulation of weak intermolecular interactions is expected to evolve as a fundamental strategy in the design of next-generation high-performance catalytic systems.
Author Contributions
X.G., Z.F., B.Z., T.X., Y.Z. and K.Z. co-wrote the manuscript. W.Z. and Z.W. made constructive comments on the article. K.Z. conceived and designed the overall framework and provided intellectual and technical guidelines. All authors discussed the results and commented on the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding Statement
This work is supported by the Beijing Natural Science Foundation (No. 2232068) and the Fundamental Research Funds for the Central Universities (2025MS158).
Acknowledgements
The authors appreciate the financial support from the Beijing Natural Science Foundation (No. 2232068).
References
2. Zhang W, Qin R, Fu G, Zheng N. Hydrogen Bond Network Induced by Surface Ligands Shifts the Semi-hydrogenation Selectivity over Palladium Catalysts. Journal of the American Chemical Society. 2023;145(18):10178–86.
3. Wang T, Zhang Y, Huang B, Cai B, Rao RR, Giordano L, et al. Enhancing oxygen reduction electrocatalysis by tuning interfacial hydrogen bonds. Nature Catalysis. 2021;4(9):753–62.
4. Li X, Lv B, Zhang X-P, Jin X, Guo K, Zhou D, et al. Introducing Water‐Network‐Assisted Proton Transfer for Boosted Electrocatalytic Hydrogen Evolution with Cobalt Corrole. Angewandte Chemie International Edition. 2022;61(9):e202114310.
5. Ren G, Zhou M, Wang H. Weakened Interfacial Hydrogen Bond Connectivity Drives Selective Photocatalytic Water Oxidation toward H2O2 at Water/Brookite-TiO2 Interface. Journal of the American Chemical Society. 2024;146(9):6084–93.
6. Qian G, Zhu L, Zeng Y, Li J, Liu J, Fei C, et al. Mechanism and Origins of Weak Bonding-Controlled Selectivities in Cinchoninium-Catalyzed Umpolung Michael Addition of Imines. CCS Chemistry. 2025;7(6):1797–811.
7. Sun R, Zhu Z, Tian N, Zhang Y, Huang H. Hydrogen Bonds and In situ Photoinduced Metallic Bi0/Ni0 Accelerating Z‐Scheme Charge Transfer of BiOBr@NiFe‐LDH for Highly Efficient Photocatalysis. Angewandte Chemie International Edition. 2024;63(41):e202408862.
8. Yu M-Y, Wu J, Yin G, Jiao F-Z, Yu Z-Z, Q J. Dynamic Regulation of Hydrogen Bonding Networks and Solvation Structures for Synergistic Solar-Thermal Desalination of Seawater and Catalytic Degradation of Organic Pollutants. Nano-Micro Letters. 2025;17(1):48.
9. Zhao K, Pang W, Jiang S, Hu C, Liu P, Cui D, et al. Operando reconstruction-induced CO2 reduction activity and selectivity for cobalt-based photocatalysis. Nano Research. 2023;16:4812–20.
10. Zhu Z, Hu W, Wu X, Zhang Q, Y Hu, Yan Q, et al. In situ self-assembled macroporous interconnected nanosheet arrays of Ni-1, 3, 5-benzenetricarboxylate metal−organic framework on Ti mesh as high-performance oxygen evolution electrodes. Journal of Colloid and Interface Science. 2023;639:274–83.
11. Hu W, Yan Q, Wang X, Lu J, He Q, zhang Q, et al. In Situ Controllably Self‐Assembled CoFe‐TDPAT Metal–Organic Framework Nanosheet Arrays on Iron Foam as Highly Efficient Bifunctional Catalytic Electrodes for Overall Water Splitting at Large Current Density. Advanced Functional Materials. 2025;35(1):2411904.
12. Raynal M, Ballester P, Vidal-Ferran A, Leeuwen PWNM. Supramolecular catalysis. Part 1: non-covalent interactions as a tool for building and modifying homogeneous catalysts. Chemical Society Reviews. 2014;43(5):1660–733.
13. Zhao Z, Liu Y, Wang Y. Weak Interaction Activates Esters: Reconciling Catalytic Activity and Turnover Contradiction by Tailored Chalcogen Bonding. Journal of the American Chemical Society. 2024;146(19):13296–305.
14. Zhang A-A, Wang Z-X, Fang Z-B, Li J-L, Liu T-F. Long‐range π–π stacking brings high electron delocalization for enhanced photocatalytic activity in hydrogen‐bonded organic framework. Angewandte Chemie. 2024;136(46):e202412777.
15. Zhao X, Liu Y. Unveiling the active structure of single nickel atom catalysis: critical roles of charge capacity and hydrogen bonding. Journal of the American Chemical Society. 2020;142(12):5773–7.
16. Jiao Y, Chen X Y, Stoddart J-F. Weak bonding strategies for achieving regio-and site-selective transformations. Chem. 2022;8(2):414–38.
17. Flór M, Wilkins D-M, de la Puente M, Laage D, Cassone G, Hassanali A, et al. Dissecting the hydrogen bond network of water: Charge transfer and nuclear quantum effects. Science. 2024;386(6726):eads4369.