Abstract
Immunosuppressive regulatory T cells (Tregs) are critical in maintaining immune tolerance and homeostasis of the immune system. Treg dysfunction leads to autoimmunity, while active Tregs in the tumor microenvironment hinders anti-tumor immune responses. As a master transcription factor of Tregs, forkhead box P3 (Foxp3) coordinates with different proteins in regulating Treg development and function. Such events are controlled by a series of post-translational modifications (PTMs). Among them, ubiquitin-dependent modifications of Foxp3 regulate degradation, nuclear localization, signal transduction, and suppressive function of the protein. Research has discovered different partners involved in the ubiquitination and deubiquitination of Foxp3. Understanding the mechanisms of these key PTMs of Foxp3 shall expand our knowledge of Tregs and pave the way for targeting Tregs in treating autoimmune diseases and cancer.
Keywords
Autoimmunity, Clinical immunology, Immune cells and organs, Immunotherapy, Tumor immunology
Introduction
Delicately, our immune system eliminates exogenous and endogenous threats and prevents harmful immune responses against the host. Regulatory T cells (Tregs) are indispensable in controlling immune responses and inducing immune tolerance; thus, immune homeostasis is maintained [1]. As a subset of CD4+ T cells, Tregs have been extensively studied for decades. They are best known for their ability to suppress immune responses, induce self-tolerance and help tumor cells escape immune surveillance [2-5]. Tregs mediate immune suppression via several mechanisms: They constitutively express cytotoxic T lymphocyte antigen-4 (CTLA-4), which competes with the costimulatory molecule CD28 for binding CD80/86 to downregulate T cell activation [6]. Tregs also highly express CD25 (IL-2 receptor α chain), depriving effector T cells of IL-2 [7]. Moreover, Tregs secrete immunosuppressive cytokines (e.g., IL-10 and IL-35) and molecules (e.g., adenosine) to inhibit effector T cell activation and function [8].
Transcription factor forkhead box protein 3 (Foxp3) is specifically expressed in Tregs and defines the Treg population [4,9]. Mutations in the Foxp3 gene cause immune dysregulation, polyendocrinopathy, enteropathy, and X-linked syndrome (IPEX) in humans and lead to fatal lymphoproliferative disorders in mice (Scurfy phenotypes) [10-12]. As a key regulator of Treg function, Foxp3 inhibits IL-2 production and upregulates CD25 and CTLA-4 expression [9,13]. Moreover, Foxp3 cooperates with a series of partners including NFAT [13], RUNX1 [14], RORα [15], Rel A, c-Rel [16], IRF4 [17], Eos [18] and HIF-1 [19] in manipulating Treg cell function. These Foxp3-mediated regulatory events are carefully controlled by different mechanisms to maintain immune homeostasis and post-translational modifications (PTMs) are one of them. Many studies have investigated the PTMS of Foxp3 protein, including phosphorylation, acetylation, methylation, glycosylation, and ubiquitination [20]. Ubiquitination is an ATP-dependent PTM that attaches ubiquitin to target proteins [21]. Ubiquitin-dependent regulation of Tregs in their function and plasticity was comprehensively reviewed in 2021 [22]; here, we shall revisit this topic and share some recent knowledge.
Ubiquitination and Deubiquitination
Ubiquitin is a 76-amino acid protein and can be initially activated by ubiquitin-activating enzymes (E1s) on the C terminus, then transiently carried by ubiquitin-conjugating enzymes (E2s) and eventually transferred to lysine residues of the target proteins by ubiquitin ligases (E3s) [23]. Monoubiquitination regulates protein-protein, protein-DNA, and protein-lipid interactions in different cellular processes [24] and polyubiquitination is often associated with protein degradation and cell signaling events [23]. Polyubiquitination chains can be linked to seven lysine residues (K6, K11, K27, K29, K33, K48, and K63) and methionine at position 1 (M1) of ubiquitin [25]. K48 and K63 polyubiquitination are best studied. K48 polyubiquitination induces degradation of target proteins by the 26S proteasome, while K63 polyubiquitination can regulate cell signaling and protein trafficking events [26]. Deubiquitination is catalyzed by deubiquitinating enzymes (DUBs), which cleave ubiquitin from ubiquitin-conjugated protein substrates; thus, DUBs regulate various critical cellular processes including TGF-β and NF-κB signaling, proteasome function, chromatin structure and endocytosis [27]. Ubiquitination and deubiquitination regulate Foxp3 stability, turnover, protein-protein interactions, localization, Treg development and function [22]. Given the roles Tregs play in autoimmunity and tumors, understanding the mechanisms of these PTMs will provide better ideas for targeting Foxp3 in treating autoimmune diseases and cancer. Here we shall discuss some widely studied ubiquitin-associated enzymes and proteins, and a summary diagram is shown below (Figure 1).
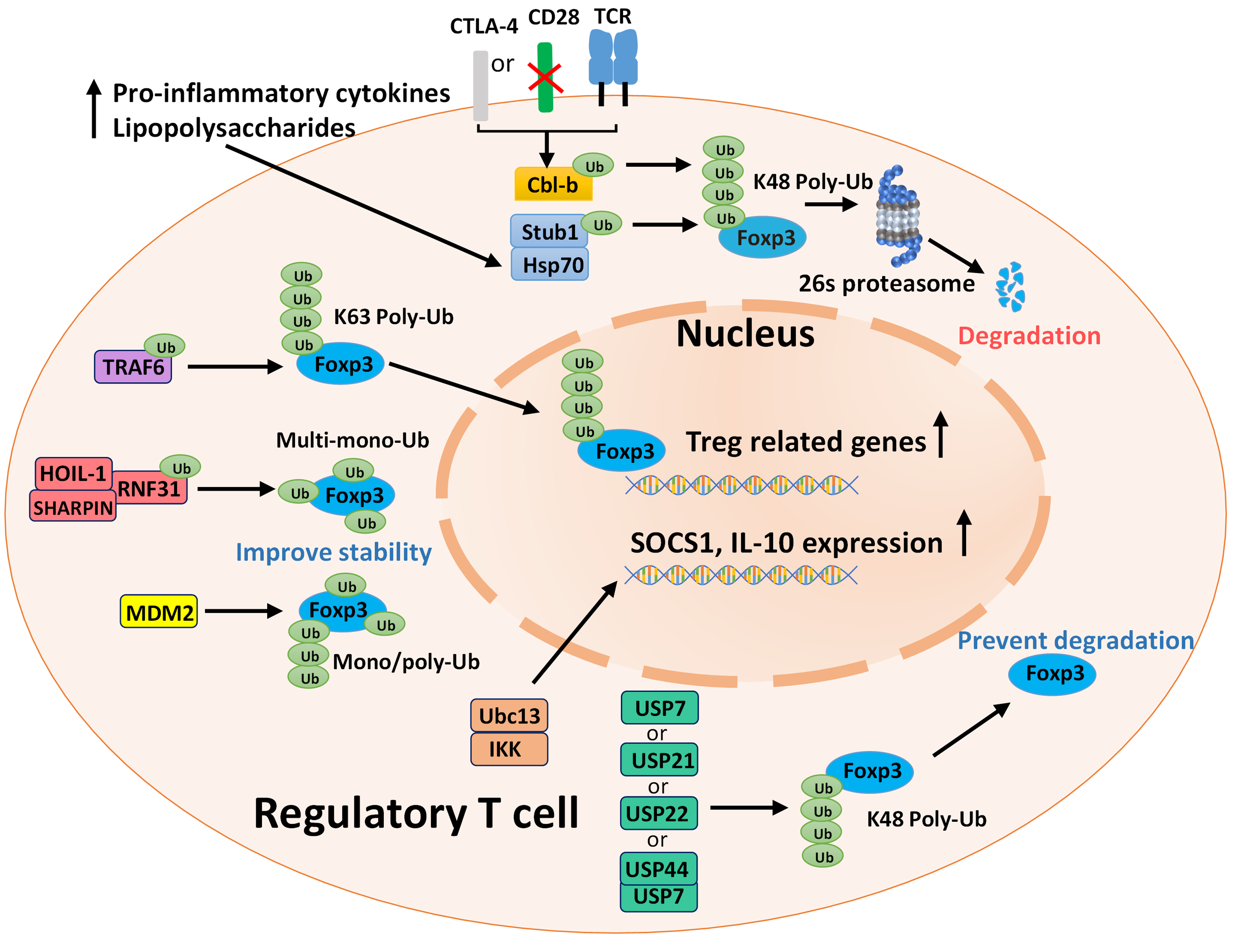
Ubiquitin Ligase-dependent Regulation of Treg Stability and Function
Stub1
STIP1 homology and U-box containing protein1 (Stub1) is a U-box-type E3 ligase required for T cell receptor (TCR)-induced NF-κB activation and IL-2 secretion [28]. Recruited by the Hsp-70 protein, Stub1 promotes Foxp3 degradation in Tregs by inducing its K48-type polyubiquitination under inflammatory stress [29]. However, such regulation can be held back. During Treg activation, Foxp3 phosphorylation induced by Nemo-like kinase (NLK) prevents the association of Stub1 and Foxp3, which subsequently stabilizes Foxp3 expression [30]. Moreover, a recent study reported that in patients with non-small-cell lung carcinoma (NSCLC), metastasis-associated lung adenocarcinoma transcript1 (MALAT1) binds the zinc finger (ZF) domain and leucine zipper (LZ) domain of Foxp3, thus masking these binding sites for Stub1 and inhibits Stub1-induced Foxp3 degradation [31]. Overall, Stub1 is a key regulator in Foxp3 stability.
TRAF6
TNF receptor-associated factor 6 (TRAF6) also directly interacts with and modifies Foxp3. This broadly expressed RING-type E3 ligase acts as an adapter molecule downstream of the IL-1 receptor, Toll-like receptor (TLR) family, and TNF receptor superfamily signaling pathways [32]. It was found that TRAF6 interacts with Foxp3 and mediates K63-type polyubiquitination at lysine 262, which is indispensable for nuclear Foxp3 localization and regulatory function [33]. Treg-specific deletion of TRAF6 abrogates Treg-mediated in vivo suppression and enhances anti-tumor responses [33]. Consistently, another group reported that when the E3 ligase of TRAF6 is inactivated, mice develop autoimmunity and inflammations in the skin, liver, and lung [34]. Treg cells expressing this E3 ligase inactive TRAF6 mutant are likely non-functional and incapable of restraining autoimmune responses in mice [34].
RNF31
The linear ubiquitin chain assembly complex (LUBAC) has three subunits, including ring finger protein 31 (RNF31/HOIP), RanBP-type and C3HC4-type zinc finger containing 1 (RBCK1/HOIL-1), and SHANK-associated RH domain interacting protein (SHARPIN/SIPL1) [35,36]. LUBAC regulates downstream signaling pathways of TCR, BCR, TNFR1, TLR, and NLRP3 [37-41]. All three LUBAC components play roles in thymic Treg development, and deletion of RNF31 results in massive loss of peripheral Tregs [42]. RNF31 is a RING-type E3 ligase, and a recent study indicates that RNF31 directly interacts with Foxp3 and stabilizes the protein via multi-monoubiquitination [43]. Moreover, upregulated RNF31 expression was observed in human gastric tumor-infiltrated Tregs, suggesting RNF31 may be a potential target for treating gastric cancer [43].
MDM2
Mouse Double Minute 2 Homolog (MDM2) is an oncogenic RING-type E3 ligase that ubiquitinates and degrades p53, a tumor suppressor [44]. Numerous studies have been focused on developing inhibitors to block MDM2-p53 interactions for cancer treatment [45]. In T cells, USP15 stabilizes MDM2, which promotes the degradation of transcription factor NFATc2 and downregulates T cell activation [46]. A later study showed that MDM2 interacts with Foxp3 and catalyzes various types of ubiquitination of Foxp3 at ten different lysine residues, stabilizing the protein [47]. These data reveal a new role of MDM2 in maintaining Foxp3 stability.
ITCH
Some other ubiquitin ligases play roles in regulating signaling pathways that indirectly mediate Treg stability and function. The Itchy E3 ubiquitin ligase (ITCH) is a HECT-type E3 ligase that plays a role in mediating allergic responses, as Th2 cells from ITCH-/- mice are resistant to anergy induction [48]. A subsequent study reported that TGF-β-mediated conversion of naïve T cells into induced Tregs (iTregs) is largely impaired in ITCH-/- mice. Further investigations confirmed that ITCH regulates Foxp3 expression by promoting ubiquitination of a transcription factor TIEG1 (TGF-β-inducible early gene 1), resulting in its nuclear translocation and transactivation of the Foxp3 promoter [49].
Cbl-b
Casitas-B-lineage lymphoma protein-b (Cbl-b) is a RING-type E3 ligase involved in regulating T cell activation thresholds and inducing immune tolerance [50]. Cbl-b deficient mice are prone to develop autoimmune diseases, and their T cells can produce IL-2 without CD28 co-stimulation [50,51]. It was discovered that in normal T cell activation, CD28 co-stimulation selectively induces Cbl-b K48-type ubiquitination and degradation by the 26S proteasome [52]. On the contrary, when T cells are activated in the absence of CD28 co-stimulation or with CTLA-4-B7 co-inhibition, Cbl-b expression is stabilized [51,53]. In terms of regulating Treg stability, Cbl-b promotes the conversion of CD4+ naïve T cells into iTregs by suppressing Akt2 of the PI3K/Akt/mTOR pathway [54]. In tTreg development, Cbl-b can be recruited by Stub1-induced Foxp3 ubiquitination. Cbl-b binds Foxp3 via its UBA domain and together with Stub1, promotes Foxp3 ubiquitination and degradation, which maintains Foxp3 at a steady state [55].
Ubc13
Ubc13 is a K63-type E2 ubiquitin-conjugating enzyme that plays a role in TNF receptor and TLR signaling pathways [56]. Treg-specific deletion of Ubc13 in mice impairs in vivo immunosuppressive function of Tregs, and Ubc13-deficient Tregs acquire effector function to produce interferon-γ (IFN-γ) and IL-17 [57]. The IκB (inhibitor of NF-κB) kinase IKK is a major downstream target of Ubc13. The Ubc-IKK signaling axis maintains Treg stability and immunosuppressive function by regulating the expression of suppressor of cytokine signaling 1 (SOCS1), a key regulator known to prevent the conversion of Tregs into Th1 or Th17-like effector T cells [58], and IL-10 [57].
A20
A20 or TNF-α induced protein 3 (TNFIP3) is a widely expressed an-inflammatory protein with both E3 ligase and DUB function, which regulates NF-κB signaling and other ubiquitin-dependent activities [59]. TRAF6 and receptor-interacting protein 1 (RIP1) are two known targets of A20 to restrict downstream NF-κB signaling [60,61]. A20 inhibits tTreg development by limiting the generation of CD4SP CD25+ GITR+ Foxp3- tTreg progenitors [62]. Moreover, A20 regulates Th17/Treg balance in rheumatoid arthritis (RA). A20 expression was found to be downregulated in the bone marrow mesenchymal stem cells (BM-MSCs) of RA patients. Overexpression of A20 in human umbilical cord MSCs (UC-MSCs), a similar cell type of BM-MSCs, is able to inhibit IL-6 expression and reverse Th17 up-regulation [63].
HIF-1
Hypoxia-inducible factor 1 (HIF-1) is a transcription factor formed from an O2-regulated HIF1-α and a HIF-1β subunit found in mammalian cells under hypoxic conditions [64]. In the hypoxic tumor microenvironment, HIF1 contributes to tumor evasion by inhibiting differentiation and function of NK and dendritic cells, mediating T cell exhaustion, and upregulating the expression of immune checkpoint molecules (e.g., PD-L1) [65]. Under normoxia, HIF-1α is both synthesized and degraded, while under hypoxia HIF-1α degradation is inhibited, resulting in its accumulation and dimerization with HIF-1β [66]. When HIF-1 is hydroxylated at prolines 402 and 562 by the prolyl hydroxylase domain protein (PHD), it binds the von Hippel-Lindau protein (VHL), which subsequently recruits the Elongin-C-Elongin-B-Cullin-2-E3-ubiquitin ligase complex and leads to HIF1 degradation [66]. Notably, HIF-1 regulates Th17/Treg balance. Under Th17-skewing conditions, HIF-1 interacts with Foxp3 and mediates their co-degradation. Meanwhile, in a STAT3-dependant manner, HIF-1 enhances Th17 differentiation by directly inducing RORγt transcription and cooperating with RORγt and p300 [19]. Consistently, it was reported that a proinflammatory cytokine IL-1β downregulates iTreg differentiation by increasing HIF-1α expression via the mTORC1 pathway [67]. Chemically stabilizing HIF-1α by dimethyloxalylglycine (DMOG) abrogates iTreg differentiation [67], and promoting HIF-1α degradation by Deltex1 (DTX1) increases Foxp3 stability [68]. Together, these data suggest HIF-1 is a potential target to regulate Treg stability.
Deubiquitination-mediated Regulation of Treg Stability and Function
USP7
Ubiquitin-specific-processing protease 7 (USP7) is a DUB found to stabilize Foxp3 expression.
van Loosdregt et al. [69] initially reported that USP7 expression is upregulated during iTreg differentiation, and USP7 interacts with and deubiquitinates Foxp3. They also showed that USP7-mediated deubiquitination prevents polyubiquitination-induced degradation of Foxp3 and improves Treg-mediated suppression in vitro and in vivo [69]. A later study showed that specific deletion of USP7 in murine Tregs triggers lethal systemic autoimmunity, and USP7 also regulates Treg function by promoting the multimerization of Tip60 and Foxp3 [70]. These data indicate that USP7 is a critical regulator of Treg function.
USP18
USP18 may also be involved in regulating Treg differentiation. USP18 is a multifunctional enzyme that negatively regulates type I, type III interferon, and NF-κB signaling [68] and DC and Th17 cell development [71,72]. A recent study revealed that USP18 deficiency enhances Treg differentiation by upregulating CD25 expression but downregulates Treg-mediated suppression [73]. Since a USP18 germline-knockout mouse model was used in this study, impacts of other USP18 deficient immune cells were not excluded. Further studies using Treg-specific USP18 knockout mice were required to further confirm the roles of USP18 in controlling Treg stability and function.
USP21
USP21 is a multifunctional DUB that regulates NF-κB, Hippo, MAPK/ERK, GATA3, Wnt, and Hedgehog signaling pathways [74]. GATA3 is a transcription factor required for Treg function [75]. An early study showed that USP21 interacts with and stabilizes GATA3, which is pivotal for Treg suppressive function, through deubiquitination [76]. Later the same group discovered that USP21 prevents Foxp3 degradation via direct deubiquitination and the generation of Th1-cell-like Tregs [77]. Thus, upon TCR activation, Foxp3 binds the USP21 gene promoter and induces its expression; in return, USP21 prevents Foxp3 degradation thus maintaining Treg stability, and forming a positive feedback loop [76, 77].
USP22
USP22 is a cancer stem cell marker and enzymatic subunit of the human SAGA transcriptional cofactor complex required for cell-cycle progression [78]. Using a CRISPR-based pooled screening method, USP22 was found to be a positive regulator of Foxp3 expression [79]. USP22 deficiency in Tregs dampens Foxp3 expression and Treg-mediated suppressive function, resulting in spontaneous autoimmunity and enhanced anti-tumor responses [79]. Moreover, E3 ubiquitin ligase RNF20 was identified as a negative regulator of Foxp3, and ablation of RNF20 was able to rescue impaired Foxp3 transcription in USP22 deficient Tregs, suggesting reciprocal regulation of Foxp3 by USP22 and RNF20 [79].
USP44
USP44 is involved in regulating cell cycle [80], stem cell differentiation [81], and tumorigenesis [82]. Recently, one study identified another role of USP44 in modulating Foxp3 expression and Treg function [83]. Initially, USP44 expression was found to be upregulated in both natural Tregs (nTregs) and iTregs. Further investigations confirmed that USP44 interacts with, deubiquitinates, and stabilizes Foxp3. Moreover, USP44 cooperates with USP7 in deubiquitinating Foxp3, while USP44 mainly targets K48-linked polyubiquitinated Foxp3. In mice, Treg-specific deletion of USP44 largely impairs their immunosuppressive function in vitro and in vivo. Notably, in three types of tumor models (MC38 colon carcinoma, B16F10 melanoma, and EL4 thymoma), Treg-specific USP44 deficiency inhibits tumor growth and enhances anti-tumor immune responses [83]. These data reveal a new USP44-dependent pathway in regulating Treg development and function and suggest USP44 be a drug target in treating autoimmune diseases and cancer.
Clinical Implication and Future Perspectives
Since the discovery of Treg cells, much attention has been drawn to elucidating the mechanisms of Treg development and Treg-mediated immune suppression. Recent research has shown the therapeutic potential of targeting Tregs to treat cancer and autoimmune diseases and prevent graft vs host disease (GvHD) in transplantation [20]. Depending on the disease settings, different Treg-target strategies are needed for better outcomes. In cancer immunotherapy, antibodies against surface molecules expressed by tumor Tregs such as CD25, CTLA-4, PD-1, GITR, 4-1BB, OX-40, LAG3, TIGIT, CCR4, and CCR8 can induce Treg-depletion in the tumor microenvironment, which enhances anti-tumor responses [5]. Adoptive transfer therapy (ACT) of Tregs has been widely used in clinical trials to prevent GvHD and treat autoinflammatory and autoimmune diseases [84]. Engineered Tregs are one of the main Treg products developed for the ACT. They express a chimeric antigen receptor (CAR) or specific TCR, which recognizes an antigen of interest. Recombinant CARs are MHC-independent with high and adjustable affinity; thus, CAR-Tregs have broader applicability than TCR-Tregs [85]. CAR-Tregs have shown their therapeutic potency in preclinical models of GvHD and autoimmune diseases, including type 1 diabetes, RA, and multiple sclerosis [86]. Overall, targeting Tregs in a wide range of diseases is promising, and yet mechanisms of their suppressive function still require further investigation for better clinical applications.
As a key regulator of Treg differentiation and function, Foxp3 forms different large protein complexes which are controlled by PTMs. The idea of targeting Foxp3 is tempting but also challenging. Transcription factors are often regulated by interactions with DNA or multiple proteins, and they lack enzymatic activity and binding pockets for small molecules [87]. Therefore, interfering PTMs of Foxp3 may be more feasible in controlling Foxp3 activity. Overactive E3 ligases induce K48-linked polyubiquitination of Foxp3 that results in abnormal degradation of Foxp3 and impaired Treg function, sequentially leading to autoimmunity. Elevated expression of Stub1 in RA patients induces the imbalance of Th17/Tregs, via non-degradative ubiquitination of aryl hydrocarbon receptor (AHR) [88]. However, in the tumor microenvironment, E3 ligases stabilizing Foxp3 or DUBs reversing its K48-linked polyubiquitination amplifies Treg-mediated immunosuppression, compromising anti-tumor immunity. Upregulated RNF31 in Tregs of gastric cancer patient is associated with decreased survival of the patients [43]. Depleting USP22 or USP44 in Tregs impairs their suppressive function and impedes tumor growth [79,83]. These findings confirm that dysregulation of Treg stability and function mediated by E3 ligases and DUBs contributes to the development of autoimmune disorders and cancer.
Research targeting E3 ligases or DUBs in murine tumor models has shed light on therapeutic applications of ubiquitin-dependent regulation of Foxp3: A small molecule TRAF6 inhibitor 6877002 can reduce the number of intratumor Tregs by limiting their migration into the tumor. P5091, a USP7 inhibitor, impairs Treg suppressive function without dampening conventional T cell responses, which improves the efficacy of anti-tumor vaccines and anti-PD1 immune checkpoint inhibitors, and anti-tumor immunity [70]. Hence, small molecule drugs which only interfere with ubiquitin-dependent PTMs of Tregs could be a good approach to control Treg function.
Conclusion
In this article, we have reviewed some key ubiquitin-associated enzymes and proteins that regulate Treg function and plasticity in T cell development, autoimmune diseases, and cancer. Ubiquitin-mediated PTMs of Foxp3 regulate the transcription factor's expression, localization, and function and subsequently influence Treg-mediated immune suppression. Recent studies have largely broadened our knowledge of these PTMs, and some promising Foxp3-associated targets have come to light. In the future, modulating ubiquitination and deubiquitination of Foxp3 are likely to be efficient and specific means to tune Treg function when in need.
Funding Statement
This work was supported by the National Key R&D Program of China (2021YFC2400500), the National Natural Science Foundation of China (Grant 32170925), Shenzhen Science and Technology Program (KQTD20210811090115019), and the startup fund of SIAT, CAS.
Conflicts of Interest
The authors declare no conflicts of interest.
References
2. Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology. 1970;18(5):723-37.
3. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155(3):1151-64.
4. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330-6.
5. Tanaka A, Sakaguchi S. Targeting Treg cells in cancer immunotherapy. Eur J Immunol. 2019;49(8):1140-6.
6. Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192(2):303-10.
7. Barthlott T, Moncrieffe H, Veldhoen M, Atkins CJ, Christensen J, O'Garra A, et al. CD25+ CD4+ T cells compete with naive CD4+ T cells for IL-2 and exploit it for the induction of IL-10 production. Int Immunol. 2005;17(3):279-88.
8. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol. 2012;3:51.
9. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057-61.
10. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20-1.
11. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27(1):68-73.
12. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27(1):18-20.
13. Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126(2):375-87.
14. Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T, et al. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007;446(7136):685-9.
15. Du J, Huang C, Zhou B, Ziegler SF. Isoform-specific inhibition of ROR alpha-mediated transcriptional activation by human FOXP3. J Immunol. 2008;180(7):4785-92.
16. Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, et al. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity. 2009;31(6):932-40.
17. Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature. 2009;458(7236):351-6.
18. Pan F, Yu H, Dang EV, Barbi J, Pan X, Grosso JF, et al. Eos mediates Foxp3-dependent gene silencing in CD4+ regulatory T cells. Science. 2009;325(5944):1142-6.
19. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146(5):772-84.
20. Dong Y, Yang C, Pan F. Post-Translational Regulations of Foxp3 in Treg Cells and Their Therapeutic Applications. Front Immunol. 2021;12:626172.
21. Ciehanover A, Hod Y, Hershko A. A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem Biophys Res Commun. 1978;81(4):1100-5.
22. Dong Y, Pan F. Ubiquitin-Dependent Regulation of Treg Function and Plasticity. Adv Exp Med Biol. 2021;1278:63-80.
23. Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503-33.
24. Magits W, Sablina AA. The regulation of the protein interaction network by monoubiquitination. Curr Opin Struct Biol. 2022;73:102333.
25. Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203-29.
26. Wang G, Gao Y, Li L, Jin G, Cai Z, Chao JI, et al. K63-linked ubiquitination in kinase activation and cancer. Front Oncol. 2012;2:5.
27. Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123(5):773-86.
28. Wang S, Li Y, Hu YH, Song R, Gao Y, Liu HY, et al. STUB1 is essential for T-cell activation by ubiquitinating CARMA1. Eur J Immunol. 2013;43(4):1034-41.
29. Chen Z, Barbi J, Bu S, Yang HY, Li Z, Gao Y, et al. The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity. 2013;39(2):272-85.
30. Fleskens V, Minutti CM, Wu X, Wei P, Pals C, McCrae J, et al. Nemo-like Kinase Drives Foxp3 Stability and Is Critical for Maintenance of Immune Tolerance by Regulatory T Cells. Cell Rep. 2019;26(13):3600-12 e6.
31. Li M, Shi M, Hu C, Chen B, Li S. MALAT1 modulated FOXP3 ubiquitination then affected GINS1 transcription and drived NSCLC proliferation. Oncogene. 2021;40(22):3870-84.
32. Kobayashi T, Walsh MC, Choi Y. The role of TRAF6 in signal transduction and the immune response. Microbes Infect. 2004;6(14):1333-8.
33. Ni X, Kou W, Gu J, Wei P, Wu X, Peng H, et al. TRAF6 directs FOXP3 localization and facilitates regulatory T-cell function through K63-linked ubiquitination. EMBO J. 2019;38(9).
34. Petrova T, Bennett K, Nanda S, Strickson S, Scudamore CL, Prescott AR, et al. Why are the phenotypes of TRAF6 knock-in and TRAF6 knock-out mice so different? PLoS One. 2022;17(2):e0263151.
35. Kirisako T, Kamei K, Murata S, Kato M, Fukumoto H, Kanie M, et al. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006;25(20):4877-87.
36. Tokunaga F, Nakagawa T, Nakahara M, Saeki Y, Taniguchi M, Sakata S, et al. SHARPIN is a component of the NF-kappaB-activating linear ubiquitin chain assembly complex. Nature. 2011;471(7340):633-6.
37. Oikawa D, Hatanaka N, Suzuki T, Tokunaga F. Cellular and Mathematical Analyses of LUBAC Involvement in T Cell Receptor-Mediated NF-kappaB Activation Pathway. Front Immunol. 2020;11:601926.
38. Sasaki Y, Sano S, Nakahara M, Murata S, Kometani K, Aiba Y, et al. Defective immune responses in mice lacking LUBAC-mediated linear ubiquitination in B cells. EMBO J. 2013;32(18):2463-76.
39. Sasaki Y, Iwai K. Crucial Role of Linear Ubiquitin Chain Assembly Complex-Mediated Inhibition of Programmed Cell Death in TLR4-Mediated B Cell Responses and B1b Cell Development. J Immunol. 2018;200(10):3438-49.
40. Rodgers MA, Bowman JW, Fujita H, Orazio N, Shi M, Liang Q, et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J Exp Med. 2014;211(7):1333-47.
41. Ning S, Luo L, Yu B, Mai D, Wang F. Structures, functions, and inhibitors of LUBAC and its related diseases. J Leukoc Biol. 2022.
42. Teh CE, Lalaoui N, Jain R, Policheni AN, Heinlein M, Alvarez-Diaz S, et al. Linear ubiquitin chain assembly complex coordinates late thymic T-cell differentiation and regulatory T-cell homeostasis. Nat Commun. 2016;7:13353.
43. Zhu F, Yi G, Liu X, Zhu F, Zhao A, Wang A, et al. Ring finger protein 31-mediated atypical ubiquitination stabilizes forkhead box P3 and thereby stimulates regulatory T-cell function. J Biol Chem. 2018;293(52):20099-111.
44. Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell. 2006;21(3):307-15.
45. Konopleva M, Martinelli G, Daver N, Papayannidis C, Wei A, Higgins B, et al. MDM2 inhibition: an important step forward in cancer therapy. Leukemia. 2020;34(11):2858-74.
46. Zou Q, Jin J, Hu H, Li HS, Romano S, Xiao Y, et al. USP15 stabilizes MDM2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nat Immunol. 2014;15(6):562-70.
47. Wang A, Yang M, Liang R, Zhu F, Zhu F, Liu X, et al. Mouse Double Minute 2 Homolog-Mediated Ubiquitination Facilitates Forkhead Box P3 Stability and Positively Modulates Human Regulatory T Cell Function. Front Immunol. 2020;11:1087.
48. Venuprasad K, Elly C, Gao M, Salek-Ardakani S, Harada Y, Luo JL, et al. Convergence of Itch-induced ubiquitination with MEKK1-JNK signaling in Th2 tolerance and airway inflammation. The J Clin Invest. 2006;116(4):1117-26.
49. Venuprasad K, Huang H, Harada Y, Elly C, Subramaniam M, Spelsberg T, et al. The E3 ubiquitin ligase Itch regulates expression of transcription factor Foxp3 and airway inflammation by enhancing the function of transcription factor TIEG1. Nat Immunol. 2008;9(3):245-53.
50. Bachmaier K, Krawczyk C, Kozieradzki I, Kong YY, Sasaki T, Oliveira-dos-Santos A, et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature. 2000;403(6766):211-6.
51. Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, Hu RJ, et al. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000;403(6766):216-20.
52. Zhang J, Bardos T, Li D, Gal I, Vermes C, Xu J, et al. Cutting edge: regulation of T cell activation threshold by CD28 costimulation through targeting Cbl-b for ubiquitination. J Immunol. 2002;169(5):2236-40.
53. Li D, Gal I, Vermes C, Alegre ML, Chong AS, Chen L, et al. Cutting edge: Cbl-b: one of the key molecules tuning CD28- and CTLA-4-mediated T cell costimulation. J Immunol. 2004;173(12):7135-9.
54. Qiao G, Zhao Y, Li Z, Tang PQ, Langdon WY, Yang T, et al. T cell activation threshold regulated by E3 ubiquitin ligase Cbl-b determines fate of inducible regulatory T cells. J Immunol. 2013;191(2):632-9.
55. Zhao Y, Guo H, Qiao G, Zucker M, Langdon WY, Zhang J. E3 Ubiquitin Ligase Cbl-b Regulates Thymic-Derived CD4+CD25+ Regulatory T Cell Development by Targeting Foxp3 for Ubiquitination. J Immunol. 2015;194(4):1639-45.
56. Fukushima T, Matsuzawa S, Kress CL, Bruey JM, Krajewska M, Lefebvre S, et al. Ubiquitin-conjugating enzyme Ubc13 is a critical component of TNF receptor-associated factor (TRAF)-mediated inflammatory responses. Proc Natl Acad Sci U S A. 2007;104(15):6371-6.
57. Chang JH, Xiao Y, Hu H, Jin J, Yu J, Zhou X, et al. Ubc13 maintains the suppressive function of regulatory T cells and prevents their conversion into effector-like T cells. Nat Immunol. 2012;13(5):481-90.
58. Takahashi R, Nishimoto S, Muto G, Sekiya T, Tamiya T, Kimura A, et al. SOCS1 is essential for regulatory T cell functions by preventing loss of Foxp3 expression as well as IFN-{gamma} and IL-17A production. J Exp Med. 2011;208(10):2055-67.
59. Malynn BA, Ma A. A20: A multifunctional tool for regulating immunity and preventing disease. Cell Immunol. 2019;340:103914.
60. Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5(10):1052-60.
61. Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430(7000):694-9.
62. Fischer JC, Otten V, Kober M, Drees C, Rosenbaum M, Schmickl M, et al. A20 Restrains Thymic Regulatory T Cell Development. J Immunol. 2017;199(7):2356-65.
63. Feng Z, Zhai Y, Zheng Z, Yang L, Luo X, Dong X, et al. Loss of A20 in BM-MSCs regulates the Th17/Treg balance in Rheumatoid Arthritis. Sci Rep. 2018;8(1):427.
64. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92(12):5510-4.
65. You L, Wu W, Wang X, Fang L, Adam V, Nepovimova E, et al. The role of hypoxia-inducible factor 1 in tumor immune evasion. Med Res Rev. 2021;41(3):1622-43.
66. Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE. 2007;2007(407):cm8.
67. Feldhoff LM, Rueda CM, Moreno-Fernandez ME, Sauer J, Jackson CM, Chougnet CA, et al. IL-1beta induced HIF-1alpha inhibits the differentiation of human FOXP3(+) T cells. Sci Rep. 2017;7(1):465.
68. Hsiao HW, Hsu TS, Liu WH, Hsieh WC, Chou TF, Wu YJ, et al. Deltex1 antagonizes HIF-1alpha and sustains the stability of regulatory T cells in vivo. Nat Commun. 2015;6:6353.
69. van Loosdregt J, Fleskens V, Fu J, Brenkman AB, Bekker CP, Pals CE, et al. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity. 2013;39(2):259-71.
70. Wang L, Kumar S, Dahiya S, Wang F, Wu J, Newick K, et al. Ubiquitin-specific Protease-7 Inhibition Impairs Tip60-dependent Foxp3+ T-regulatory Cell Function and Promotes Antitumor Immunity. EBioMedicine. 2016;13:99-112.
71. Cong XL, Lo MC, Reuter BA, Yan M, Fan JB, Zhang DE. Usp18 promotes conventional CD11b+ dendritic cell development. J Immunol. 2012;188(10):4776-81.
72. Liu X, Li H, Zhong B, Blonska M, Gorjestani S, Yan M, et al. USP18 inhibits NF-?B and NFAT activation during Th17 differentiation by deubiquitinating the TAK1�TAB1 complex. J Exp Med. 2013;210(8):1575-90.
73. Yang L, Jing Y, Kang D, Jiang P, Li N, Zhou X, et al. Ubiquitin-specific peptidase 18 regulates the differentiation and function of Treg cells. Genes Dis. 2021;8(3):344-52.
74. An T, Lu Y, Yan X, Hou J. Insights Into the Properties, Biological Functions, and Regulation of USP21. Front Pharmacol. 2022;13:944089.
75. Wang Y, Su MA, Wan YY. An essential role of the transcription factor GATA-3 for the function of regulatory T cells. Immunity. 2011;35(3):337-48.
76. Zhang J, Chen C, Hou X, Gao Y, Lin F, Yang J, et al. Identification of the E3 deubiquitinase ubiquitin-specific peptidase 21 (USP21) as a positive regulator of the transcription factor GATA3. The J Biol Chem. 2013;288(13):9373-82.
77. Li Y, Lu Y, Wang S, Han Z, Zhu F, Ni Y, et al. USP21 prevents the generation of T-helper-1-like Treg cells. Nat Commun. 2016;7:13559.
78. Zhang XY, Varthi M, Sykes SM, Phillips C, Warzecha C, Zhu W, et al. The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol Cell. 2008;29(1):102-11.
79. Cortez JT, Montauti E, Shifrut E, Gatchalian J, Zhang Y, Shaked O, et al. CRISPR screen in regulatory T cells reveals modulators of Foxp3. Nature. 2020;582(7812):416-20.
80. Stegmeier F, Rape M, Draviam VM, Nalepa G, Sowa ME, Ang XL, et al. Anaphase initiation is regulated by antagonistic ubiquitination and deubiquitination activities. Nature. 2007;446(7138):876-81.
81. Fuchs G, Shema E, Vesterman R, Kotler E, Wolchinsky Z, Wilder S, et al. RNF20 and USP44 regulate stem cell differentiation by modulating H2B monoubiquitylation. Mol Cell. 2012;46(5):662-73.
82. Zhang Y, Foreman O, Wigle DA, Kosari F, Vasmatzis G, Salisbury JL, et al. USP44 regulates centrosome positioning to prevent aneuploidy and suppress tumorigenesis. J Clin Invest. 2012;122(12):4362-74.
83. Yang J, Wei P, Barbi J, Huang Q, Yang E, Bai Y, et al. The deubiquitinase USP44 promotes Treg function during inflammation by preventing FOXP3 degradation. EMBO Rep. 2020;21(9):e50308.
84. Esensten JH, Muller YD, Bluestone JA, Tang Q. Regulatory T-cell therapy for autoimmune and autoinflammatory diseases: The next frontier. J Allergy Clin Immunol. 2018;142(6):1710-8.
85. Raffin C, Vo LT, Bluestone JA. Treg cell-based therapies: challenges and perspectives. Nat Rev Immunol. 2020;20(3):158-72.
86. Arjomandnejad M, Kopec AL, Keeler AM. CAR-T Regulatory (CAR-Treg) Cells: Engineering and Applications. Biomedicines. 2022;10(2).
87. Chen A, Koehler AN. Transcription Factor Inhibition: Lessons Learned and Emerging Targets. Trends Mol Med. 2020;26(5):508-18.
88. Wang W, Xiang T, Yang Y, Wang Z, Xie J. E3 ubiquitin ligases STUB1/CHIP contributes to the Th17/Treg imbalance via the ubiquitination of aryl hydrocarbon receptor in rheumatoid arthritis. Clin Exp Immunol. 2022;209(3):280-90.