Abstract
SARS-Cov-2 is the virus that causes the disease COVID-19. While most patients who contract the disease experience mild symptoms, a significant percentage has severe symptoms, sometimes leading to death. In this paper, the immune response to SARS-Cov-2 infection is reviewed, highlighting differences between patients with severe disease and patients with mild disease. Findings suggest that, with severe disease, there is an exaggerated inflammatory reaction to the virus while both the humoral (B cell) and cytotoxic (NK-cell and CD8+ T cell) arms of the immune system are weak and delayed. Possible treatment options are also explored. Studies in other settings have shown that IL-15 supports the cytotoxic arm of the immune response. IL-21 has been shown to support both the cytotoxic and humoral arms of the immune response. In addition, in some settings, IL-21 has been shown to actually decrease the production of IL-6 and TNF-α, reducing the inflammatory proteins involved in the cytokine storm. In other settings, the combination of IL-15 and IL-21 has been shown to be more effective than either interleukin alone in promoting an effective immune response. So if the combination of IL-15 and IL-21 is given early in the course of the disease to patients with early predictors of severe disease, the devastating effects of the infection may be avoided. Therefore, a clinical trial of these interleukins for SARS-Cov-2 disease is warranted.
Keywords
COVID-19, SARS-Cov-2, Immune cells, Interleukin-15, Interleukin-21, Immune Treatment, Severe vs. Mild COVID-19 Disease
Introduction
SARS-Cov-2 is the virus that causes the disease COVID-19. The disease has led to the worst worldwide health crisis in 100 years. While many patients remain asymptomatic, most patients develop a mild respiratory infection. Symptoms include cough, fever and chills, fatigue and body aches, headache and loss of the sense of taste or smell. However, a proportion of patients develop severe disease. Symptoms of severe disease include higher fever, hypoxia and respiratory distress, leading to hospitalization, and sometimes the need for mechanical ventilation and ultimately death [1].
The SARS-CoV-2 virus enters host cells by binding to the angiotensin-converting enzyme 2 (ACE2) receptor, a molecule present on many cells in the body at various levels of expression. The virus first invades nasal epithelial cells by binding to ACE2 [2]. There it undergoes local replication and propagation. At this stage the patient is generally asymptomatic. The immune response during this phase is limited and the patient may be highly infectious.
The virus then migrates from the nasal epithelium to the upper respiratory tract. At this stage symptoms of fever, malaise and dry cough are often present. The chemokine ligand CXCL-10 and interferons are released from the virus-infected cells. For most patients, the immune response is sufficient to contain the spread of infection at this point.
However, SARS-Cov-2 virus-infected cells have been shown to block the production of Type I IFNs by several mechanisms, sometimes inhibiting an effective antiviral immune response [3]. Approximately 20% of the patients progress to involvement of the lower respiratory tract. There, the virus invades type II alveolar epithelial cells via the same host receptor, ACE2, which is highly expressed in these cells. The virus then undergoes replication in the cells of the alveoli, infecting both type II and adjacent type I alveolar cells as well as resident macrophages. In addition to inhibiting IFNs, it has been shown that the SARS-Cov-2 spike 1 protein induces the NK cell inhibitory protein, NKG2A, resulting in reduced NK cell effectiveness [4]. Inhibitory markers PD-1 and Tim-3, additional markers of lymphocyte exhaustion, are also induced by the virus [5]. In the presence of a weakened cellular immune response, the virus-infected pneumocytes now release many different cytokines including IL-1, IL-6, IL-8, TNF-α, CXCL-10, monocyte chemoattractant protein-1(MCP-1), macrophage inflammatory protein-1α (MIP-1α) and interferons, resulting in the cytokine storm. Subsequently IL-8 recruits neutrophils to the site of infection. CXCL10 induces NK cells to the site of infection. MCP-1 and MIP-1alpha recruit monocytes, macrophages and other inflammatory cells to the site of infection. Interferons activate NK cells and phagocytes. Sequestration of these inflammatory cells in lung tissue, as well as continuing viral replication in adjacent healthy cells ultimately leads to diffuse alveolar damage and acute respiratory distress syndrome, hallmarks of severe disease. Furthermore, TNF-α, IL-1 and IL-6, known as acute phase proteins, produce fever. TNF-α also stimulates the expression of adhesion molecules, inducing the extravasation of monocytes and neutrophils. TNF-α also stimulates endothelial cells to express proteins that induce a blood clot, a process that normally serves as an aid in preventing pathogens from entering the bloodstream. However, in the setting of cytokine storm, TNF-α can also cause vasodilation, leading to an increase in vascular permeability, and contributing to blood clotting in small vessels throughout the body in a process known as disseminated intravascular coagulation (DIC). This can ultimately lead to the failure of vital organs such as kidneys, liver, heart and lungs.
Given the variable immune responses patients have to the infection, an early intervention that promotes an effective immune response could be transformative. In order to understand the variable responses, the immune response to viruses in general [6], and this virus in particular, will be reviewed. This review will highlight features of the immune response that differ in patients with severe disease vs. mild disease. Following this review, possible treatment options will be explored.
The Innate Immune Response
As with the immune response to pathogens in general, the immune response to viruses occurs in two phases, the innate immune response and the adaptive immune response. The innate immune response occurs within hours of the infection. The adaptive immune response starts to be effective 4 to 7 days after exposure to the virus. Both phases of the immune response are complex and involve many molecular mediators, intracellular processes and cell surface receptors. Those features that have differential expression in severe vs. mild COVID-19 disease will be emphasized in this review (Figure 1).
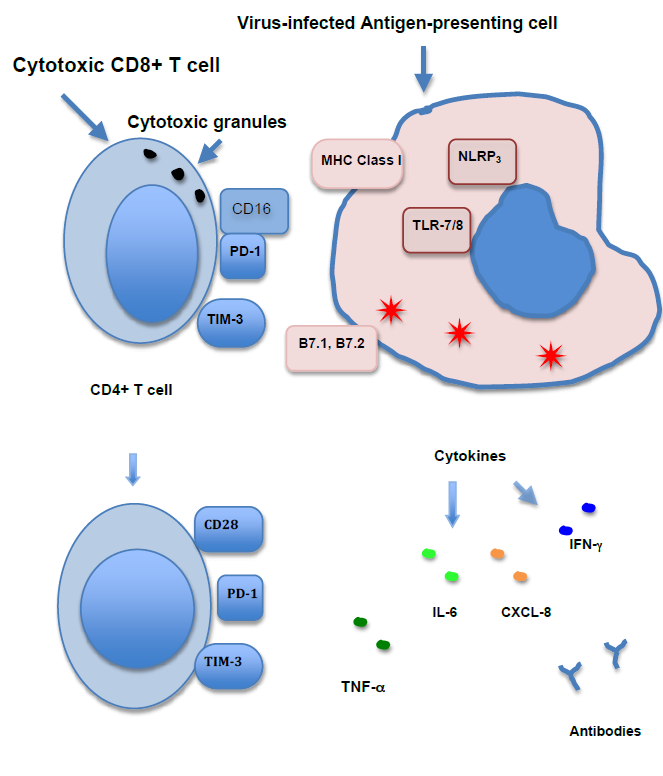
The immune system is initially activated when the virus infects the innate sensor cells, macrophages and dendritic cells. Toll-like receptors (TLR) in these cells detect the presence of the virus’s single-stranded RNA in their cytoplasm as a PAMP (pathogen-associated molecular pattern). This detection is non-specific. It does not depend on the particular sequence of bases in the RNA, only the presence of single-stranded RNA in endosomes in the cytoplasm. TLR-7 and TLR-8 recognize single-stranded RNA.
The TLRs then activate the transcription factor NFκB which in turn induces the production of pro-inflammatory cytokines TNF-α and IL-6. Activated macrophages also release the chemokine CXCL8 (IL-8), which attracts neutrophils to the site of infection. TNF-α performs several functions, including activation of complement, opsonization, neutrophil mobilization to sites of infection and an increase in body temperature; this creates an environment for decreased viral replication and an increased immune response. Results of this activity include fever, increased blood vessel dilatation and an increase in neutrophil adhesion molecules on endothelial cells.
Similar to TLRs, NOD-like receptors (NLRs) can also activate the NLRP3 signaling pathway, generating proinflammatory cytokines through the formation of a multiprotein complex known as the inflammasome. This activation occurs in the presence of DAMPs (Damage- Associated Molecular Patterns) from cell remnants as infected cells die. This mechanism has been shown to be especially prominent in the elderly and obese [7].
TLRs also induce the expression of anti-viral cytokines, including type 1 interferon (IFN). Type 1 IFNs are important mediators in the defense against viral infections, increasing the number of Major Histocompatibility complex (MHC) class I molecules that are present on nearly all cells. The increased expression of MHC class I molecules strengthens the response of cytotoxic effector cells to infection. Type I IFNs also act to induce a state of resistance to viral replication in all cells by suppressing the translation of viral RNA. IFNs also activate MHC class II molecules and recruit lymphocytes to the site of infection.
Virus-infected innate sensor cells also release IL-12 which activates NK cells. NK cells then produce IFN-γ and TNF-α. NK cells also cytotoxically kill infected cells in response to nonspecific PAMPS without the presence of specific antigens. They do this by releasing the cytotoxic granules perforin, which creates pores in the infected cells, and granzyme, which passes through the pores into the cells to kill them. NK cells also exhibit Fc receptors (CD16) and act as the effector cells for antibody-dependent cellmediated cytotoxicity (ADCC) by detecting the Fc portions of cell-adherent antibodies that are generated during the adaptive immune.
All of the cells of the immune system, including NK cells, are also supported in their growth by interleukins. Released by various cells of the body to support the growth and differentiation of immune cells in response to infection, interleukins usually work in concert with other interleukins and cytokines to produce a balanced immune response. One such interleukin, Interleukin 15 (IL-15), is involved in the stimulation and growth of NK cells in the course of the immune response.
By inducing the inflammatory response and activating cytotoxic NK cells, the innate immune response tends to keep the viral infection in check until the adaptive immune response can be activated and effectively eliminate the infection.
The Adaptive Immune Response
The adaptive immune system targets the infection by recognizing specific antigens on the virus. Two of the products of the adaptive immune system are cytotoxic CD8+ T cells that directly kill infected cells and B cells that produce and release antibodies specific to the virus. The antibodies perform several functions, all leading to the elimination of the virus.
The adaptive immune response is initiated by the interaction of antigen-presenting cells and CD4+ T cells. An antigen-presenting cell with viral peptides bound to a cleft in the MHC class II molecule on the cell surface adheres to a CD4-T cell receptor complex on a CD4+ T cell. Two co-stimulatory molecules, B7.1 and B7.2, are also induced on the cell surface of the antigen-presenting cell, prompted by TLR activation. These are required, along with the antigen-MHC complex, to activate the adaptive immune response. B7.1 and B7.2 bind to CD28 on naïve T cells. CD28 promotes T cell proliferation, cytokine production and cell survival and is necessary for optimal clonal expansion of naïve T cells. Cytokines are also released into the milieu by the antigen-presenting cells, directing the T cell to differentiate into the particular type of T cell required for its effector function. Later, as the infection subsides, T cells express inhibitory molecules to regulate and diminish the immune response. Two of these inhibitory molecules are PD-1 and Tim-3.
Activated CD4+ T cells are involved in several arms of the adaptive immune response. They strengthen the B cell’s response when it encounters a complementary antigen, produce IFN-γ and IL-2, a general growth factor for T cells, and promote effector CD8+ T cell differentiation and CD8+ memory cells, leading to long-term immunity.
When a cell is infected by a virus, viral peptides are presented on MHC class I molecules. CD8+ T cells interact with these virus peptide-MHC class I complex molecules on the cell’s surface. The CD8+ T cells become cytotoxic, killing the infected cell by releasing cytotoxic granules at the surface of the target cell, similar to the action of NK cells. CD8+ cytotoxic T cells also release the cytokines INF-γ, TNF-α and LT-α, which contribute to host defense. As with NK cells, the same interleukin, IL-15, also supports the growth and enhances the survival of long-term memory CD8+ T cells.
During the development of the person’s immune system, a vast number of B cells are formed, each carrying a unique antibody on its surface. When a B cell encounters a pathogen that expresses a complementary antigen to that particular B cell’s antibody, the B cell is activated. B cells also receive help from CD4+ T cells as mentioned above. B cells that react with the initially detected antigen migrate to germinal centers in the lymph nodes, where their antibodies hyper-mutate into antibodies with high affinity for the antigen in a process known as affinity maturation. As they proliferate, B cells expressing these antibodies also undergo class switching from IgM to IgG or IgA class in the process of developing a more effective antibody response. At the end of this process, the adaptive immune response includes a strong, high-affinity highly specific IgG antibody response, which is generally very effective in combating the infection. Some of the B cells then differentiate into plasma cells that produce large quantities of these antibodies to be released into the bloodstream and at sites of infection. In germinal centers, Interleukin-21 (IL-21) stimulates the proliferation of B cells. IL-21 also supports antibody class switching to IgG1, IgG3 and IgA. IgA works predominately in the mucosal immune system, preventing viruses from attaching to epithelial cells. Both IgA and IgG serve as neutralizing antibodies. IL-21 also supports the differentiation and growth of antibody-producing plasma cells.
High-affinity antibodies attack viruses in four ways. The first way is by neutralizing viruses. They attach to the virus, blocking the virus from infecting host cells. These antibody-virus complexes are then vulnerable to degradation by scavenger immune cells, such as macrophages. Antibodies also cause the viruses to clump together in a process called agglutination, increasing their susceptibility to phagocytosis. Antibodies also interfere with viral infection because the constant portion of the antibody (Fc) that is adherent to an infected cell is detected by an NK cell. While NK cells are thought of as part of the innate immune system, the NK cells cytotoxically kill the infected cells in response to antibodies. Antibodies also activate complement. Complement factors attached to the virus also make the virus more vulnerable to phagocytosis.
The SARS-COV-2 Virus Counters the Immune Response
Studies of COVID-19 infection that examined the differences between the immune response that occurred in patients with severe versus mild symptoms [8,9] show that hallmarks of severe disease include a cytokine storm, a decreased absolute B cell count and/or decreased NK cell and CD8+ T cell counts. The cytokine storm includes elevation of IL-6, IL-10 and TNF-α [10]. There is evidence that this elevated inflammatory response may be related to activation of the inflammasome by NLRP3, which is associated with aging [7,11,12]. Also, IFN-γ production tends to be lower in severe disease. CD28 is also decreased on CD4+ and CD8+ T cells in severe disease [5]. The same study shows that the expression of inhibitory markers PD-1 and Tim-3 on CD4+ and CD8+ T cells is increased in severe disease. Although the time course of the antibody response in mild disease versus severe cases has not been completely elucidated, one study shows that the ratio of IgG antibodies targeting the spike (S) protein and host cell receptor binding domain (RBD) on the virus over antibodies targeting the nucleocapsid antigen is lower in severe cases compared with mild cases [13]. In another study, generation of S and RBD specific IgG’s occurred one week later in patients with severe Covid-19 disease compared with patients with mild disease. [14]. This study also shows that these antibodies are 2-fold higher in recovered patients who now test RNA-negative for the virus compared with those who are still RNA-positive. Additionally, there is evidence [15] that patients with the severe disease do not mount a strong IgG response against the spike proteins on the virus. In yet another study, there is evidence that the IgG response overall is weaker in patients with the critical disease compared with patients with the mild disease [16]. Most recently, a study shows that neutrophil activation markers, including CXCL8 (IL-8) predict severe disease [17]. It has also been shown that patients with mild disease have interferon secreting T cells present in the blood early in the course of the disease while patients with the severe disease do not exhibit this feature [18]. In keeping with this finding, there is evidence that this SARS-Cov-2 virus specifically antagonizes the interferon response during active infection [3].
In addition, there is evidence that a coordinated immune response, consisting of both CD4+ and CD8+ T cells along with an antibody response, is more effective than an immune response from a single arm of the immune system. [19].
So, in severe disease, the acute phase reactants TNF-α and IL-6 and neutrophil activation markers are elevated while IFN-γ is lower. NK cells and cytotoxic T cells tend to be lower. B cells also tend to be lower, and there is a delay in the production of high-affinity antibodies (Figure 2).
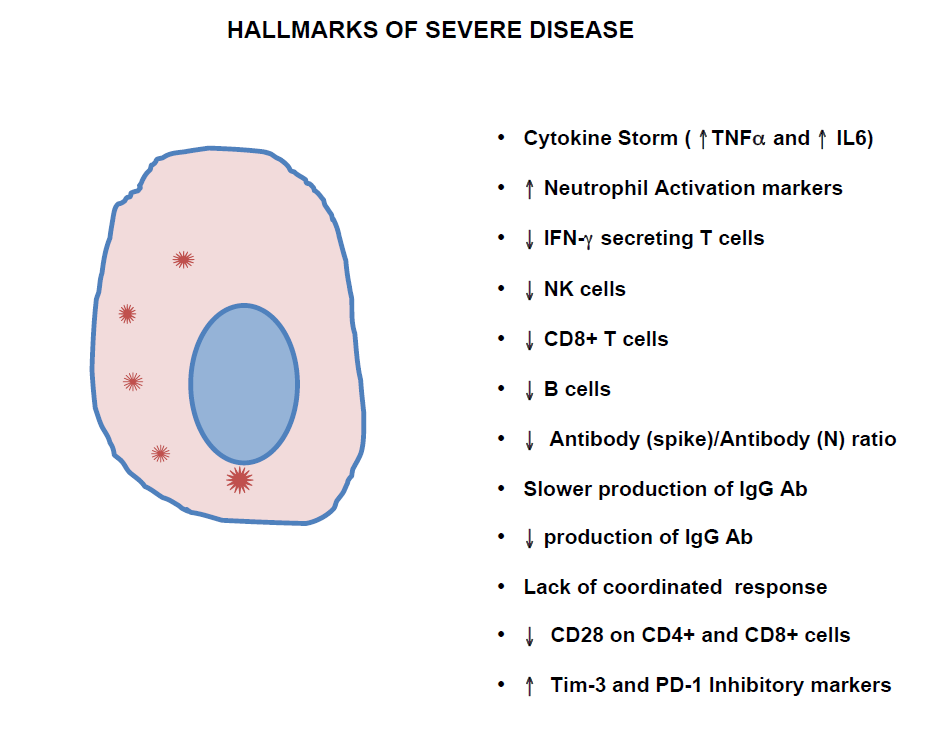
Overall, these findings suggest that there is an exaggerated inflammatory reaction to the virus while both the humoral (B cell) and cytotoxic (NK-cell and CD8+ T cell) arms of the immune system are weak in patients with severe disease. A plausible explanation for the severity of the disease in those with risk factors is that the cellular and antibody immune response to eliminate the virus is delayed, so that by the time the cytotoxic effects become prominent, the virally-infected cell load is so large that it stimulates DAMP induced inflammasomes, activating a cytokine storm [7]. Thus an intervention that promotes a cellular response of cytotoxic cells and the differentiation and production of high-affinity antibodies early in the disease may be helpful in promoting an effective immune response.
Treatments
While some treatments currently do exist, none are transformative. Both Remdesivir [20], a drug that acts as a nucleoside analogue and blocks RNA synthesis by the virus, and immunosuppressive steroids [21] are somewhat helpful in patients that already have severe disease.
Also, monoclonal antibodies have shown some efficacy [22,23]. Monoclonal antibody therapy is generally considered to be a form of passive immunotherapy. However, the presence of antibodies that contain Fc segments bound to the virus is a powerful stimulus for the induction of the adaptive immune response. So monoclonal antibody treatment may in fact promote the development of an adaptive immune response as well as serve in the process of passive immunity. Research on this question is currently suggesting that this may be the case [24].
Actions of Various Interleukins
As noted above, treatments that specifically strengthen cells of the humoral and cytotoxic responses may be very helpful. In the past, there have been multiple studies of the effects of giving recombinant interleukins designed to strengthen these two arms of the immune response in cancer patients [25-32]. IL-2, IL-15 and IL-21, while all sharing the same cytokine receptor gamma subunit, have been shown to elicit different immunological responses in vitro and in vivo. IL-2 has been shown to activate effector and memory CD8+ T cells, promote the growth of CD4 T cells and NK cells and support the suppression of the T cell immune response by stimulating CD4+ Treg cells [33,34]. However, in previous studies, IL-2 treatment has led to capillary leak syndrome as well as increased proinflammatory cytokines (cytokine storm) [35].
IL-15 has been shown to stimulate cytotoxic CD8+ T-cell and NK cell responses, and promote memory-phenotype CD8+ T cells [25,26,27,36]. Of note, IL-15 can support the proliferation of T cells that have diminished expression of CD28, and enable their interaction with antigenpresenting cells [37]. As noted above, CD28 expression is diminished during severe Covid-19 infection.
IL-21 has been shown to co-stimulate T and NK cell proliferation and function, promote the transition from an innate to an adaptive response, promote B cell differentiation into plasma cells, up-regulate IgG1 and IgG3 antibody formation and counteract suppression by CD4+ regulatory T cells [30,31,38-41]. IL-21 has also been shown to reverse the effects of inhibitory markers PD-1 and Tim-3, at least on the innate cytotoxic NK cells [42]. As noted above, these markers are increased on CD8+ cytotoxic cells in patients with severe disease.
While problematic side effects have been identified in these studies, they occur in the relatively long course of treatment required for cancer therapy. The side effects may not occur in the short time frame required to treat an acute viral infection.
A Combination of Interleukins May Be Warranted
Regarding the current pandemic, treatment of COVID-19 infection with IL-2 is currently in clinical trials. However, as noted above, in previous studies, IL-2 treatment has led to capillary leak syndrome as well as increased proinflammatory cytokines (cytokine storm) [35]. Given that cytokine storm is a major complication of the infection, exogenous IL-2 may be contraindicated. In contrast, IL- 21 does not have this profile of complicating side effects. In fact, IL-21 has been shown to decrease IL-6 and TNFalpha production [43]. Treatment with IL-15 also does not lead to the capillary leak syndrome [33], and is known to strengthen the body’s cytotoxic response, augmenting antibody-dependent cell-mediated cytotoxicity [26]. Furthermore, IL-15 may also have a supportive effect of inducing memory lymphocytes after the initial infection, so as to induce long-term immunity to the coronavirus [35,44]. IL-21, along with IL-4, has been shown to support the differentiation and proliferation of B cells [31].
In vivo, these cytokines generally do not act in an isolated fashion, but multiple interleukins act in concert. For instance, the combination of IL-15 and IL-21 has been shown to be more effective than either interleukin alone in promoting an effective immune response [44-46]. IL- 21 has been shown to support sustaining the proliferation and homing of CD8+ effector cells while CD-15 supports CD8+ cell cytotoxic activation [34]. IL-21 also supports B cell and subsequent plasma cell function, supporting the generation of antibodies, and counters suppression of the immune response by CD4+ Treg cells [39,40].
Others have urged the initiation of a clinical trial that examines the use of IL-15 for patients that present with diminished NK cell or CD8+ T cell counts [47]. But given the known effects of IL-21 in promoting CD8 effector cell proliferation as well as B cell and plasma cell development, and the finding that the combination of IL-15 and IL-21 may be more effective than IL-15 alone, I would also urge the addition of IL-21 to IL-15 in one arm of the study, particularly in patients that present with either diminished B cell counts [48] or elevated neutrophil activation markers as early predictors of severe disease [13].
Conclusions
Studies suggest that, compared to patients with mild disease, there is an exaggerated inflammatory reaction and weakened humoral (B cell) and cytotoxic (NK-cell and CD8+ T cell) immune responses in patients with severe disease. In other settings, the combination of Interleukin-15 and Interleukin-21 has been shown to effectively support these weakened arms of the immune response. A clinical trial of these interleukins for SARS-Cov-2 infection is warranted. Severe disease may be avoided by directing the immune response to arms of the immune system that are known to be most effective in treating the disease, at an early stage of the disease.
Conflict of Interest
The author declares no conflicts of interest.
References
2. Mason RJ. Thoughts on the alveolar phase of COVID-19. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2020 Jul 1;319(1):L115-L120.
3. Xia H, Cao Z, Xie X, Zhang X, Chen JY, Wang H, et al. Evasion of Type I Interferon by SARS-CoV-2. Cell Reports. 2020 10 6;33(1):108234.
4. Bortolotti D, Gentili V, Rizzo S, Rotola A, Rizzo R. SARS-CoV-2 Spike 1 Protein Controls Natural Killer Cell Activation via the HLA-E/NKG2A Pathway. Cells. 2020 08 26;9(9):E1975.
5. Wang F, Hou H, Luo Y, Tang G, Wu S, Huang M, et al. The laboratory tests and host immunity of COVID-19 patients with different severity of illness. JCI Insight. 2020 05 21;5(10):137799.
6. Murphy K, Weaver C. Janeway’s immunobiology. 9th Edition, New York (NY) Norton, W. W. & Company, Inc; 2016.
7. Yang A, Guduguntla LS, Yang B. Potentials of Interferons and Hydroxychloroquine for the Prophylaxis and Early Treatment of COVID-19. Journal of Cellular Immunology. 2020;2(6):333-40.
8. Zhang Y, Wang X, Li X, Xi D, Mao R, Wu X, et al. Potential contribution of increased soluble IL-2R to lymphopenia in COVID-19 patients. Cellular & Molecular Immunology. 2020 08;17(8):878-80.
9. Tan M, Liu Y, Zhou R, Deng X, Li F, Liang K, et al. Immunopathological characteristics of coronavirus disease 2019 cases in Guangzhou, China. Immunology. 2020 07;160(3):261-8.
10. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. Journal of Clinical Investigation. 2020 05 1;130(5):2620-9.
11. Mueller AL, McNamara MS, Sinclair DA. Why does COVID-19 disproportionately affect older people. Aging (Albany NY). 2020 05 29;12(10):9959-81.
12. Youm YH, Kanneganti TD, Vandanmagsar B, Zhu X, Ravussin A, Adijiang A, et al. The Nlrp3 inflammasome promotes age-related thymic demise and immunosenescence. Cell Reports. 2012 Jan 26;1(1):56-68.
13. Röltgen K, Powell AE, Wirz OF, Stevens BA, Hogan CA, Najeeb J, et al. Defining the features and duration of antibody responses to SARS-CoV-2 infection associated with disease severity and outcome. Science Immunology. 2020 12 7;5(54):eabe0240.
14. Li K, Huang B, Wu M, Zhong A, Li L, Cai Y, et al. Dynamic changes in anti-SARS-CoV-2 antibodies during SARS-CoV-2 infection and recovery from COVID-19. Nature Communications. 2020 11 27;11(1):6044.
15. Sun B, Feng Y, Mo X, Zheng P, Wang Q, Li P, et al. Kinetics of SARS-CoV-2 specific IgM and IgG responses in COVID-19 patients. Emerging Microbes & Infections. 2020 Dec;9(1):940-8.
16. Hou H, Wang T, Zhang B, Luo Y, Mao L, Wang F, et al. Detection of IgM and IgG antibodies in patients with coronavirus disease 2019. Clinical & Translational Immunology. 2020 May;9(5):e01136.
17. Meizlish ML, Pine AB, Bishai JD, Goshua G, Nadelmann ER, Simonov M, et al. A neutrophil activation signature predicts critical illness and mortality in COVID-19. Blood Advances. 2021 03 9;5(5):1164-77.
18. Tan AT, Linster M, Tan CW, Le Bert N, Chia WN, Kunasegaran K, et al. Early induction of functional SARSCoV- 2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Reports. 2021 02 9;34(6):108728.
19. Rydyznski Moderbacher C, Ramirez SI, Dan JM, Grifoni A, Hastie KM, Weiskopf D, et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell. 2020 11 12;183(4):996-1012.e19.
20. Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the Treatment of Covid-19 - Final Report. The New England Journal of Medicine. 2020 11 5;383(19):1813-26.
21. Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L, et al. Dexamethasone in Hospitalized Patients with Covid-19. The New England Journal of Medicine. 2021 Feb 25;384(8):693-704.
22. Chen P, Nirula A, Heller B, Gottlieb RL, Boscia J, Morris J, et al. SARS-CoV-2 Neutralizing Antibody LYCoV555 in Outpatients with Covid-19. The New England Journal of Medicine. 2021 01 21;384(3):229-37.
23. Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, et al. REGN-COV2, a Neutralizing Antibody Cocktail, in Outpatients with Covid-19. The New England Journal of Medicine. 2021 01 21;384(3):238-51.
24. Pelegrin M, Naranjo-Gomez M, Piechaczyk M. Antiviral Monoclonal Antibodies: Can They Be More Than Simple Neutralizing Agents. Trends in Microbiology. 2015 Oct;23(10):653-65.
25. Robinson TO, Schluns KS. The potential and promise of IL-15 in immuno-oncogenic therapies. Immunology Letters. 2017 10;190:159-68.
26. Knudson KM, Hodge JW, Schlom J, Gameiro SR. Rationale for IL-15 superagonists in cancer immunotherapy. Expert Opinion on Biological Therapy. 2020 Jul 2;20(7):705-9.
27. Romee R, Cooley S, Berrien-Elliott MM, Westervelt P, Verneris MR, Wagner JE, et al. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT- 803 to treat relapse after transplantation. Blood. 2018 06 7;131(23):2515-27.
28. Steele N, Anthony A, Saunders M, Esmarck B, Ehrnrooth E, Kristjansen PE, et al. A phase 1 trial of recombinant human IL-21 in combination with cetuximab in patients with metastatic colorectal cancer. British Journal of Cancer. 2012 Feb 28;106(5):793-8.
29. Schmidt H, Brown J, Mouritzen U, Selby P, Fode K, Svane IM, et al. Safety and Clinical Effect of Subcutaneous Human Interleukin-21 in Patients with Metastatic Melanoma or Renal Cell Carcinoma: A Phase I Trial. Clinical Cancer Research. 2010 Nov 1;16(21):5312-9.
30. Zarkavelis G, Kefas A, Pentheroudakis G. The emerging role of Interleukin-21 as an antineoplastic immunomodulatory treatment option. Translational Cancer Research. 2017 Mar;6(S2):S328-S330.
31. Croce M, Rigo V, Ferrini S. IL-21: a pleiotropic cytokine with potential applications in oncology. Journal of Immunology Research. 2015;2015:696578.
32. Davis ID, Brady B, Kefford RF, Millward M, Cebon J, Skrumsager BK, et al. Clinical and Biological Efficacy of Recombinant Human Interleukin-21 in Patients with Stage IV Malignant Melanoma without Prior Treatment: A Phase IIa Trial. Clinical Cancer Research. 2009 Mar 15;15(6):2123-9.
33. Conlon KC, Miljkovic MD, Waldmann TA. Cytokines in the Treatment of Cancer. Journal of Interferon & Cytokine Research. 2019 01;39(1):6-21.
34. Hinrichs CS, Spolski R, Paulos CM, Gattinoni L, Kerstann KW, Palmer DC, et al. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood. 2008 Jun 1;111(11):5326- 33.
35. Dutcher JP, Schwartzentruber DJ, Kaufman HL, Agarwala SS, Tarhini AA, Lowder JN, et al. High dose interleukin-2 (Aldesleukin) - expert consensus on best management practices-2014. Journal of Immunotherapy Cancer. 2014 Sep 16;2(1):26.
36. Zhang X, Sun S, Hwang I, Tough DF, Sprent J. Potent and Selective Stimulation of Memory-Phenotype CD8+ T Cells In Vivo by IL-15. Immunity. 1998 May;8(5):591-9.
37. Perera PY, Lichy JH, Waldmann TA, Perera LP. The role of interleukin-15 in inflammation and immune responses to infection: implications for its therapeutic use. Microbes and Infection. 2012 Mar;14(3):247-61.
38. Kasaian MT, Whitters MJ, Carter LL, Lowe LD, Jussif JM, Deng B, et al. IL-21 Limits NK Cell Responses and Promotes Antigen-Specific T Cell Activation. Immunity. 2002 Apr;16(4):559-69.
39. Kim-Schulze S, Kim HS, Fan Q, Kim DW, Kaufman HL. Local IL-21 promotes the therapeutic activity of effector T cells by decreasing regulatory T cells within the tumor microenvironment. Molecular Therapy. 2009 Feb;17(2):380-8.
40. Ozaki K. A Critical Role for IL-21 in Regulating Immunoglobulin Production. Science. 2002 Nov 22;298(5598):1630-4.
41. Peluso I, Fantini MC, Fina D, Caruso R, Boirivant M, MacDonald TT, et al. IL-21 Counteracts the Regulatory T Cell-Mediated Suppression of Human CD4+ T Lymphocytes. Journal of Immunology. 2007 Jan 15;178(2):732-9.
42. Seo H, Jeon I, Kim BS, Park M, Bae EA, Song B, et al. IL-21-mediated reversal of NK cell exhaustion facilitates anti-tumour immunity in MHC class I-deficient tumours. Nature Communications. 2017 06 6;8:15776.
43. Li SN, Wang W, Fu SP, Wang JF, Liu HM, Xie SS, et al. IL-21 modulates release of proinflammatory cytokines in LPS-stimulated macrophages through distinct signaling pathways. Mediators of Inflammation. 2013;2013:548073.
44. Zeng R, Spolski R, Finkelstein SE, Oh S, Kovanen PE, Hinrichs CS, et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. Journal of Experimental Medicine. 2005 Jan 3;201(1):139-48.
45. Kishida T, Asada H, Itokawa Y, Cui F, Shin-Ya M, Gojo S, et al. Interleukin (IL)-21 and IL-15 genetic transfer synergistically augments therapeutic antitumor immunity and promotes regression of metastatic lymphoma. Molecular Therapy. 2003 Oct;8(4):552-8.
46. de Rham C, Ferrari-Lacraz S, Jendly S, Schneiter G, Dayer JM, Villard J. The proinflammatory cytokines IL-2, IL-15 and IL-21 modulate the repertoire of mature human natural killer cell receptors. Arthritis Research & Therapy. 2007;9(6):R125.
47. Kandikattu HK, Venkateshaiah SU, Kumar S, Mishra A. IL-15 immunotherapy is a viable strategy for COVID-19. Cytokine & Growth Factor Reviews. 2020 08;54:24-31.
48. Wilz SW. A clinical trial of IL-15 and IL-21 combination therapy for COVID-19 is warranted. Cytokine & Growth Factor Reviews. 2021 04;58:49-50.