Abstract
Emtricitabine (FTC) is an antiviral medication designed to diminish the presence of HIV in the body, thereby impeding or preventing harm to the immune system and the onset of AIDS-related illnesses. The precise mechanisms underlying emtricitabine's inhibition of HIV-1 replication in pre-exposure prophylaxis (PrEP) are not fully comprehended. This investigation delves into the impact of emtricitabine treatments in vitro, utilizing the susceptible Jurkat cell line. Employing a sensitive real-time PCR assay and Western blotting analysis, we observed that emtricitabine pre-treatment effectively impedes HIV-1 replication. Additionally, when the treatment is discontinued, viral replication is reactivated through the recruitment of host transcription factors, such as NF-κB p65, NFAT, Ap-1, and Sp-1. Both pre-treatment and post-treatment with emtricitabine demonstrated the ability to reduce HIV production through various cellular signaling pathways. These include the attenuation of TCR-related, PI3K, and MAPK-mediated pathways, the downregulation of p-TEFb pathways responsible for transcription elongation, the obstruction of TLR signaling pathways affecting the host immune response, and the inhibition of Jak-Stat pathways associated with viral enhancement. Discontinuation of emtricitabine treatments reactivated HIV-1 replication by upregulating these cellular signaling pathways. The data strongly suggest that long-term PrEP offers continuous protection against HIV, presenting itself as a viable option for individuals consistently at high risk of infection. This ensures a steadfast and dependable form of protection.
Keywords
HIV-1, Replication, ART, PrEP, Emtricitabine, Latency, TCR
Introduction
Human immunodeficiency virus type 1 (HIV-1) is the causative agent of acquired immunodeficiency syndrome (AIDS), the advanced stage of HIV infection characterized by severe immune system damage, rendering individuals susceptible to opportunistic infections and specific cancers. Since the onset of the epidemic, approximately 85.6 million individuals have contracted the HIV virus, and approximately 40.4 million people have lost their lives due to HIV-related complications [1]. Globally, as of the conclusion of 2022, an estimated 39.0 million people were living with HIV [1]. According to existing literature, the anticipated ten and twelve-year survival probabilities following HIV diagnosis leading to AIDS onset are 26% and 19%, respectively [2].
Antiretroviral therapy (ART) stands as the established treatment for HIV, with the primary goal of impeding virus progression and managing associated symptoms. Among those with AIDS who receive ART, a 61% survival rate is expected over ten years following the development of AIDS, while those without ART have an anticipated survival rate of 18% over six years after AIDS onset [2].
HIV-1 infection and replication involve intricate interactions with various cellular signaling pathways within host immune cells, particularly CD4+ T lymphocytes (T cells) and macrophages [3]. The HIV-1 long terminal repeat (LTR) encompasses diverse consensus sequences, including NF-κB (nuclear factor κB), NFAT (nuclear factor of activated T cells), Ap-1 (activator protein 1), and Sp-1 (specificity protein 1). These host transcription factors are crucial for HIV-1 replication [4]. The binding of HIV-1 to CD4 on the surface of CD4+ T cells can initiate intracellular signaling events, often activating the T cell receptor (TCR) signaling pathways [5], including TCRα/β [6], CD3 [5], CD8 [5], PI3K/Akt [7], and/or MAPKs (p38 and Erk) [8,9]-mediated pathways, which plays pivotal role in both T cell activation and HIV-1 replication. Additionally, the HIV-1 protein Tat (trans-activator of transcription) plays a crucial role in viral transcription by interacting with host cellular factors, including Cyclin T1 and CDK9, to facilitate efficient transcription from the viral promoter in the HIV-1 LTR [3].
Pre-exposure prophylaxis (PrEP) stands as a preventive strategy to reduce the risk of HIV-1 infection among individuals at high risk of exposure to the virus. PrEP involves the proactive use of medication before potential exposure occurs [10]. The primary medication for PrEP typically combines two antiretroviral drugs, usually tenofovir disoproxil fumarate (TDF) and emtricitabine (FTC), often marketed together as Truvada and Descovy [11]. These active ingredients effectively inhibit HIV replication in the body. When taken consistently and correctly, PrEP substantially diminishes the risk of HIV transmission during sexual intercourse or other high-risk behaviors. PrEP reduces the risk of acquiring HIV through sexual contact by approximately 99% and through injection drug use by at least 74% [10].
Emtricitabine, approved by FDA (Food and Drug Administration) in 2004, is a nucleoside reverse transcriptase inhibitor (NRTI) [11]. It works by inhibiting the action of the reverse transcriptase enzyme, which is essential for the replication of HIV. By interfering with this process, emtricitabine helps to prevent the virus from multiplying in the body [11]. Beyond its inhibition of reverse transcriptase, it remains unclear whether emtricitabine affects cellular gene expression, innate immunity, or cell signaling in infected cells—factors that contribute to HIV-1 infection and replication in vitro. In this study, we report that emtricitabine pre-treatment (FTCpt) can inhibit HIV-1 replication by activating several key cellular signaling pathways. These include the downregulation of host transcription factors such as AP-1, NFAT, NF-κB, and Sp1, as well as the inhibition of T cell receptor (TCR)-related pathways that regulate transcription factors essential for HIV-1 replication. Additionally, FTCpt disrupts the p-TEFb, toll-like receptor (TLR), and JAK-STAT pathways, all of which are associated with viral replication and the establishment of HIV-1 latency.
Materials and Methods
Chemicals and reagents
Emtricitabine (FTC) was sourced from Millipore Sigma (Rockville, MD). All other chemicals were obtained from Sigma (St. Louis, MO).
Cell culture and treatments
Jurkat, clone E6-1, cells were obtained from the National Institutes of Health AIDS Research Reference and Reagent Program (Germantown, MD) and cultured at 37°C in 5% CO2 in RPMI 1640 medium containing 10% fetal calf serum, 2 mM glutamine, 50 μg/ml penicillin, and 50 μg/ml streptomycin. For HIV-1 infection and emtricitabine (FTC) treatment, cells were initially seeded at a density of 2 × 105 cells/ml for 24 hours. Subsequently, as illustrated in the Supplementary Figure 1, the cells were divided into two groups.
In the first group, on day 0, the cells were cultured for 3 days, followed by infection with known amounts of HIV-1 (MN, 109 copies per 106 cells) for 2 hours. They were then washed twice with PBS and incubated in fresh medium for an additional 4 days. On day 7, these cells were further divided into two subgroups: (0/0) - cells incubated for another 3 days, and (0/E) - cells cultured in the presence of 10 nM of emtricitabine for an additional 3 days. All cells were harvested on day 10 (Supplementary Figure 1).
In another group, on day 0, the cells were cultured in the presence of 10 nM of emtricitabine for 3 days. Subsequently, they were infected with known amounts of HIV-1 (MN, 109 copies per 106 cells) for 2 hours. Following this, they were washed twice with PBS and incubated in fresh medium with 10 nM of emtricitabine for an additional 4 days. On day 7, the cells in this group were placed in fresh medium and divided into two subgroups: (E/0) - cells incubated in the absence of emtricitabine for another 3 days, and (E/E) - cells cultured in the presence of 10 nM of emtricitabine for an additional 3 days. All cells were harvested on day 10 (Supplementary Figure 1).
RNA isolation and real-time PCR
The RNA isolation and real-time PCR were performed according to our previous reports [8,12,13]. Briefly, viral RNA was extracted from 140 µl of culture supernatant using the QIAamp Viral RNA Mini Kit. Real-time RT-PCR was performed using known concentrations of HIV-1 (MN) viral RNA as templates for standard curve generation. Primers and TaqMan probes for gag p24 were designed, and quantitative RT-PCR was conducted in a TaqMan 7500 Analyzer.
Western blot analysis
The Western blotting was performed according to our previous reports [8,12,13]. Briefly, proteins isolated from Jurkat cells were subjected to SDS-PAGE, blotted onto polyvinylidene difluoride membranes, and analyzed using antibodies against various proteins. Antibodies from Abcam (Waltham, MA), including rabbit polyclonal/mouse monoclonal antibodies against Jak1, Jak2, NF-κB p65, PLCγ-1, PKC, TCRα, and TLR1, were obtained. Additionally, mouse monoclonal antibodies against IRAK4, IRF3, and IRF7 were acquired from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit polyclonal antibodies against Akt, AP-1, Brd4, CD3, CDK9, Cyclin T1, Erk1/2, MyD88, NFAT, p38, PCAF, PI3K, p-Rpb-CTD, Stat1, Stat3, Stat5, Sp-1, TF, GAPDH, and ZAP70 were purchased from Cell Signaling Technology, Inc (Danvers, MA). The relative quantitation of protein expression was determined using ImageJ (Image Processing and Analysis in Java) from NIH website (https://imagej.nih.gov/ij/).
The cells were cultured and treated in 30 ml of medium, and three independent experiments were performed to obtain the Western blotting results.
Statistical analysis
The unpaired Student’s t test was used for data analyses as indicated, and p-value <0.05 (*) and p-value <0.01 (**) were considered significant and very significant, respectively.
Results
The EC50 (effective concentration 50) is a key metric for evaluating drug activity in cell culture systems. It represents the concentration of a drug required to inhibit viral replication by 50% in a specific cell line. For emtricitabine, EC50 values in cell cultures have been reported to range from 1.3 to 640 nM (0.0003–0.158 µg/mL) [14]. Under our experimental conditions, we observed that 10 nM of emtricitabine produced a more favorable cellular response (Supplementary Figure 2).
Emtricitabine (Figure 1A), often used in combination with other antiretroviral drugs for HIV-1 treatment, inhibits the enzyme reverse transcriptase crucial for viral replication. This interference reduces viral load, slows disease progression, and enhances immune function [10,11].
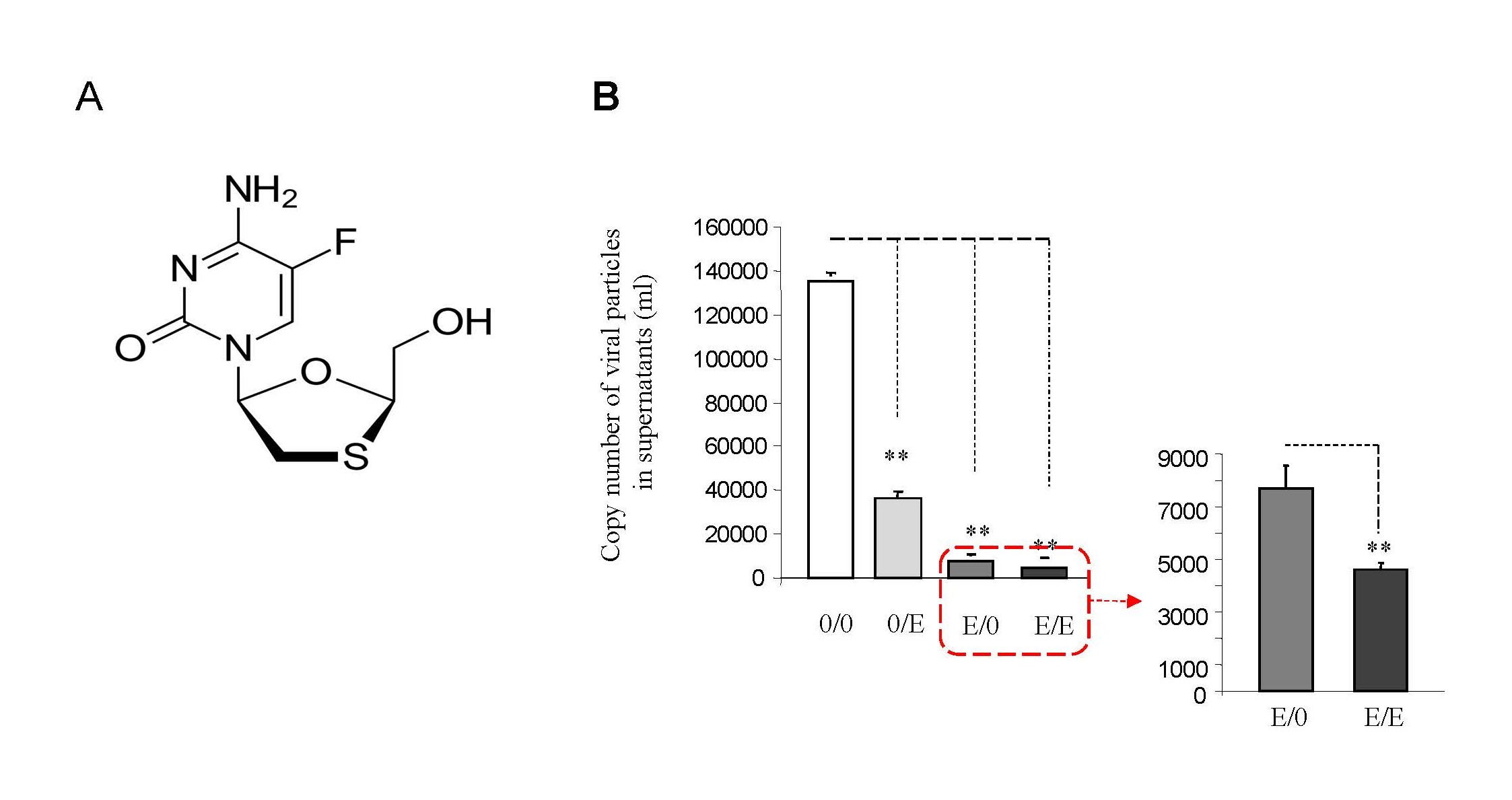
Figure 1. Emtricitabine pre-treatment inhibited HIV-1 replication with decreased viral loads. (A) The structure of emtricitabine. (B) Jurkat cells were treated with 10 nM of emtricitabine or/and infected with HIV-1, as described in the Materials and Methods section and visually depicted in the Supplementary Figure 1. A total of 140 µl of culture supernatants containing HIV-1 particles was used to isolate viral RNA for RT-PCR to assess viral replication.
FTCpt reduces the activity of host transcription factors
Given its role in HIV PrEP, we investigated if emtricitabine pre-treatment could inhibit HIV-1 replication. In vitro studies, detailed in Supplementary Figure 1, revealed significant inhibition of HIV-1 replication with emtricitabine pre-treatment (Figure 1B: panel 0/0 and 0/E vs. E/0 and E/E). Post-treatment also blocked replication, while discontinuation led to reactivation (E/0 vs. E/E). This suggests that emtricitabine pre-treatment can inhibit replication, with cessation potentially reactivating HIV-1 in Jurkat cells.
Host transcription factors, including NF-κB p65, Ap-1, Sp-1, and NFAT, crucial for HIV-1 replication [4], were examined. Western blot analysis (Figure 2) showed emtricitabine pre-treatment downregulated these factors (panel 0/0 and 0/E vs. E/0 and E/E); while emtricitabine post-treatment resulted in a significant decrease in the expression of NF-κB p65, Ap-1, Sp-1, and NFAT (as depicted in Figure 2, comparing panel 0/0 to panel 0/E and panel E/0 to panel E/E); indicating that emtricitabine inhibits replication by suppressing these key factors.
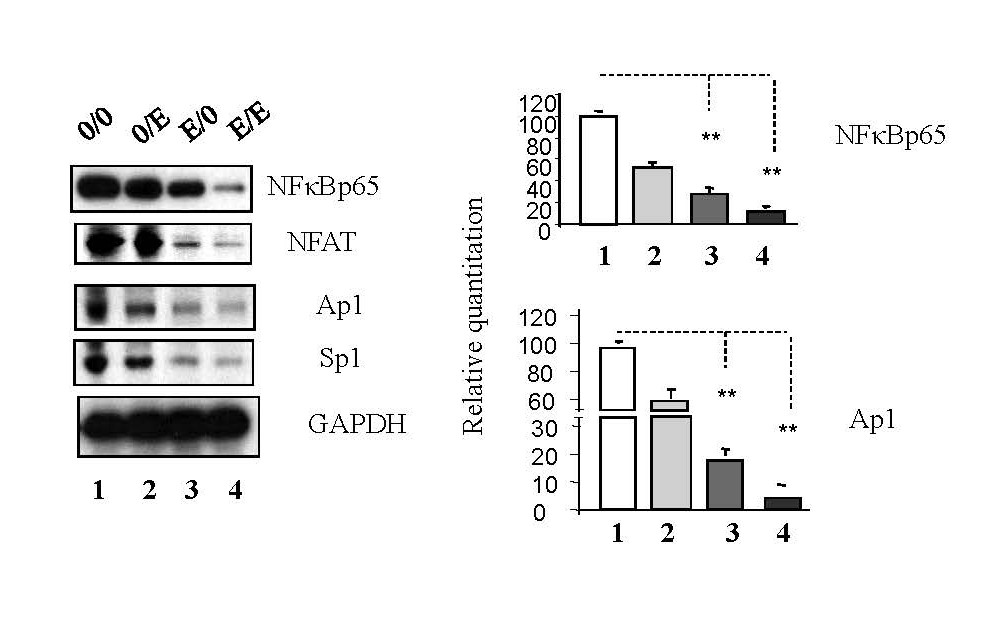
Figure 2. Emtricitabine pre-treatment inhibited activation of host transcription factors. Jurkat cells were treated with 10 nM of emtricitabine or/and infected with HIV-1, as described in the Materials and Methods section and visually depicted in the Supplementary Figure 1. Total cell lysates were subjected to Western blot analysis to detect Ap-1, NFAT, NF-κB p65, and Sp-1.
FTCpt hinders TCR-related pathways
T cells, vital in recognizing HIV-1-infected cells, rely on the activation of TCR-related pathways. Emtricitabine pre-treatment led to downregulation of TCRα, CD3, ZAP-70, PLCγ-1, and PKC (Figure 3A, seen in panel 0/0 and panel 0/E compared to panel E/0 and panel E/E) in TCR pathways. At the same time, emtricitabine post-treatment resulted in a significant decrease in the expression of TCRα, CD3, ZAP-70, PLC-γ1, and PKC (as shown in Figure 3A, comparing panel 0/0 to panel 0/E and panel E/0 to panel E/E).
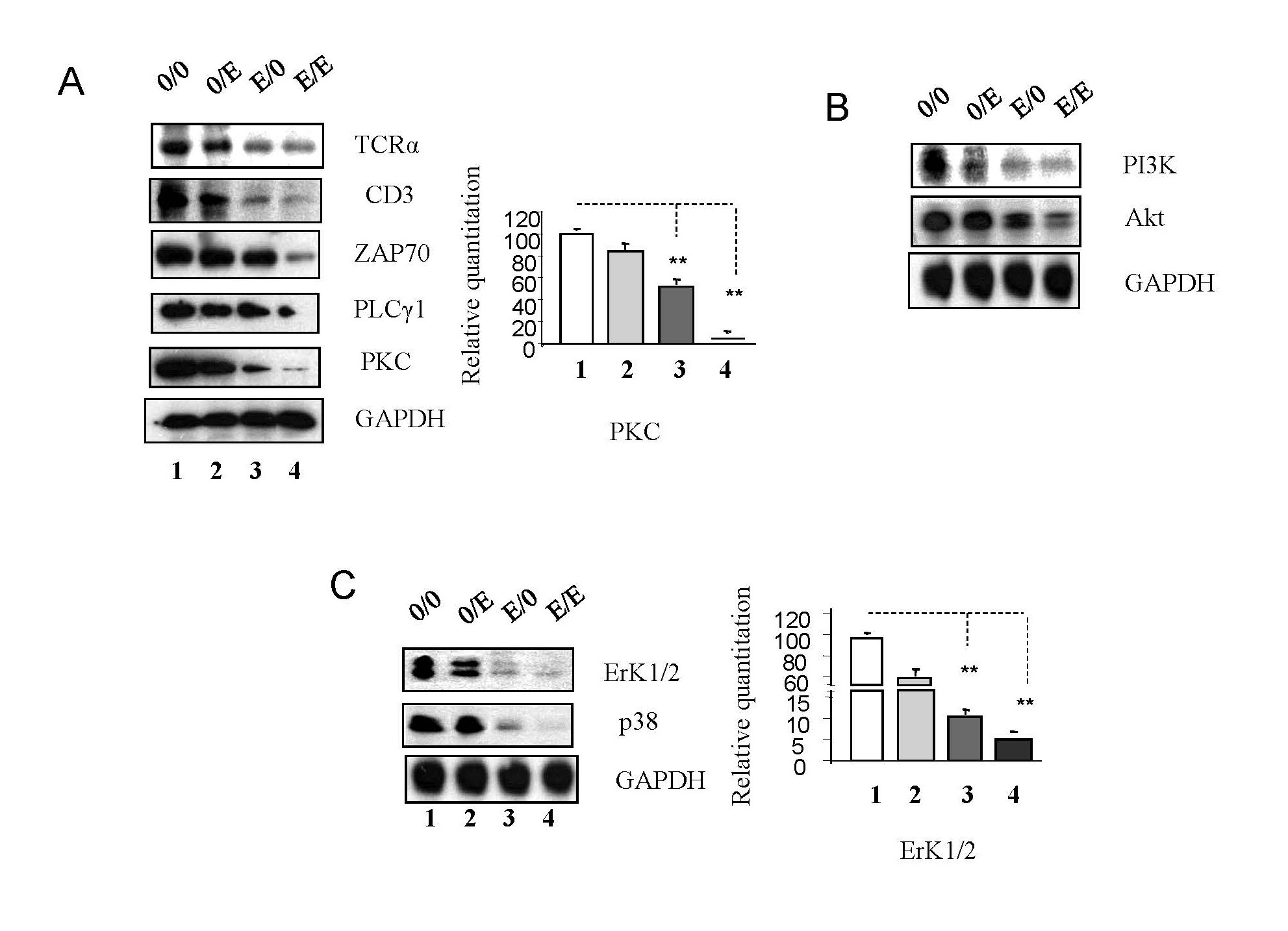
Figure 3. Emtricitabine pre-treatment decreased activation of TCR-related signaling pathways. Jurkat cells were treated with 10 nM of emtricitabine or/and infected with HIV-1, as described in the Materials and Methods section and visually depicted in the Supplementary Figure 1. Total cell lysates were subjected to Western blot analysis to detect (A) CD3, TCRα, PKC, PLCγ1 and ZAP 70; (B) PI3K and Akt; and (C) ERK1/2 and p38.
PI3K/Akt pathway components and MAPKs (Erk1/2 and p38) were downregulated by emtricitabine pre-treatment (as shown in panel 0/0 and panel 0/E compared to panel E/0 and panel E/E) (Figures 3B and 3C). Simultaneously, emtricitabine post-treatment significantly decreased the expression of PI3K and Akt, Erk1/2 and p38 (as demonstrated in Figures 3B and 3C, comparing panel 0/0 to panel 0/E and panel E/0 to panel E/E); suggesting that emtricitabine inhibits TCR-related pathways crucial for host cell activation. The cessation of emtricitabine treatments can reactivate these pathways, potentially contributing to viral resurgence.
These data indicate that emtricitabine pre-treatment inhibits HIV-1 replication by suppressing host transcription factors and TCR-related pathways. The cessation of treatment may lead to pathway reactivation, emphasizing the importance of consistent PrEP for sustained protection against HIV-1 infection.
FTCpt inhibits the activation of Tat-mediated recruitment of transcriptional machinery
HIV-1 Tat, a key regulatory protein, plays a pivotal role in viral transcription and replication [15]. Tat interacts with the histone acetyltransferase PCAF (p300/CBP-associated factor), crucial for viral replication and transcription [16]. Emtricitabine pre-treatment downregulated Tat and PCAF expression, potentially hindering histone acetylation and limiting access to the transcriptional machinery (Figure 4A).
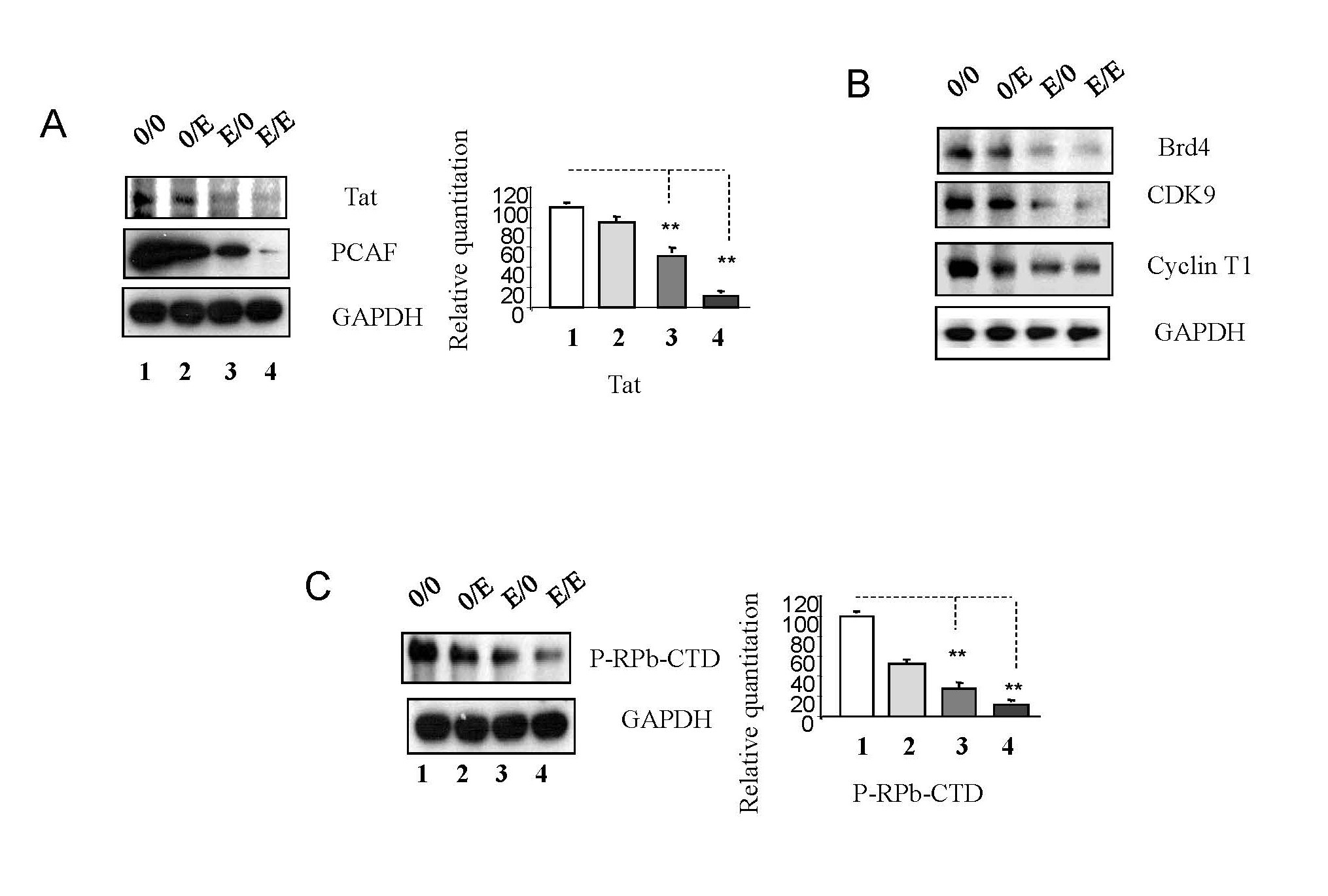
Figure 4. Emtricitabine pre-treatment inhibited activation of that Tat-mediated recruitment of transcriptional machinery on HIV-1 replication. Jurkat cells were treated with 10 nM of emtricitabine or/and infected with HIV-1, as described in the Materials and Methods section and visually depicted in the Supplementary Figure 1. Total cell lysates were subjected to Western blot analysis to detect (A) Tat and PCAF; (B) Brd4, CDK9 and Cyclin T1; (C) P-RPb-CTD.
Tat binds to TAR (Transactivation response element) RNA structure at the 5’ end of the nascent viral RNA [15,16], recruiting Brd4 (Bromodomain-containing protein 4) [17], an essential factor in gene expression regulation. Emtricitabine pre-treatment led to the downregulation of Brd4, CDK9, and Cyclin T1, components of the p-TEFb complex [15,16,18] (Figure 4B). This complex facilitates RNA polymerase II progression along viral RNA, promoting efficient viral gene expression [18,19]. Additionally, emtricitabine inhibited the phosphorylation of p-RPb-CTD, a critical step in transcriptional elongation (4C), in panel 0/0 and panel 0/E compared to panel E/0 and panel E/E (Figure 3C). Meanwhile, emtricitabine post-treatment significantly reduced the expression of Brd4, CDK9, and Cyclin T1 (Figure 4B), as well as p-RPb-CTD (Figure 4C), in panel 0/0 compared to panel 0/E and in panel E/0 compared to panel E/E.
These results indicate that emtricitabine pre-treatment may reduce Tat activity, hinder histone acetylation via PCAF downregulation, and block the p-TEFb pathway. This inhibition suggests a potential mechanism for limiting viral transcription and replication.
FTCpt blocks TLR pathways
The Toll-like receptor (TLR) pathway, crucial for immune responses [20,21], was targeted by emtricitabine pre-treatment. Expression of key TLR pathway molecules, including TLR1, MyD88, IRAK4, IRF7, and IRF3, was significantly reduced. Emtricitabine post-treatment continued to suppress these components, suggesting a sustained impact on the TLR pathway (Figures 5A–5D), suggesting that emtricitabine pre-treatment inhibits TLR pathways, a vital component of the immune response to HIV-1. The virus's evasion strategies underscore the significance of these pathways in infection control.
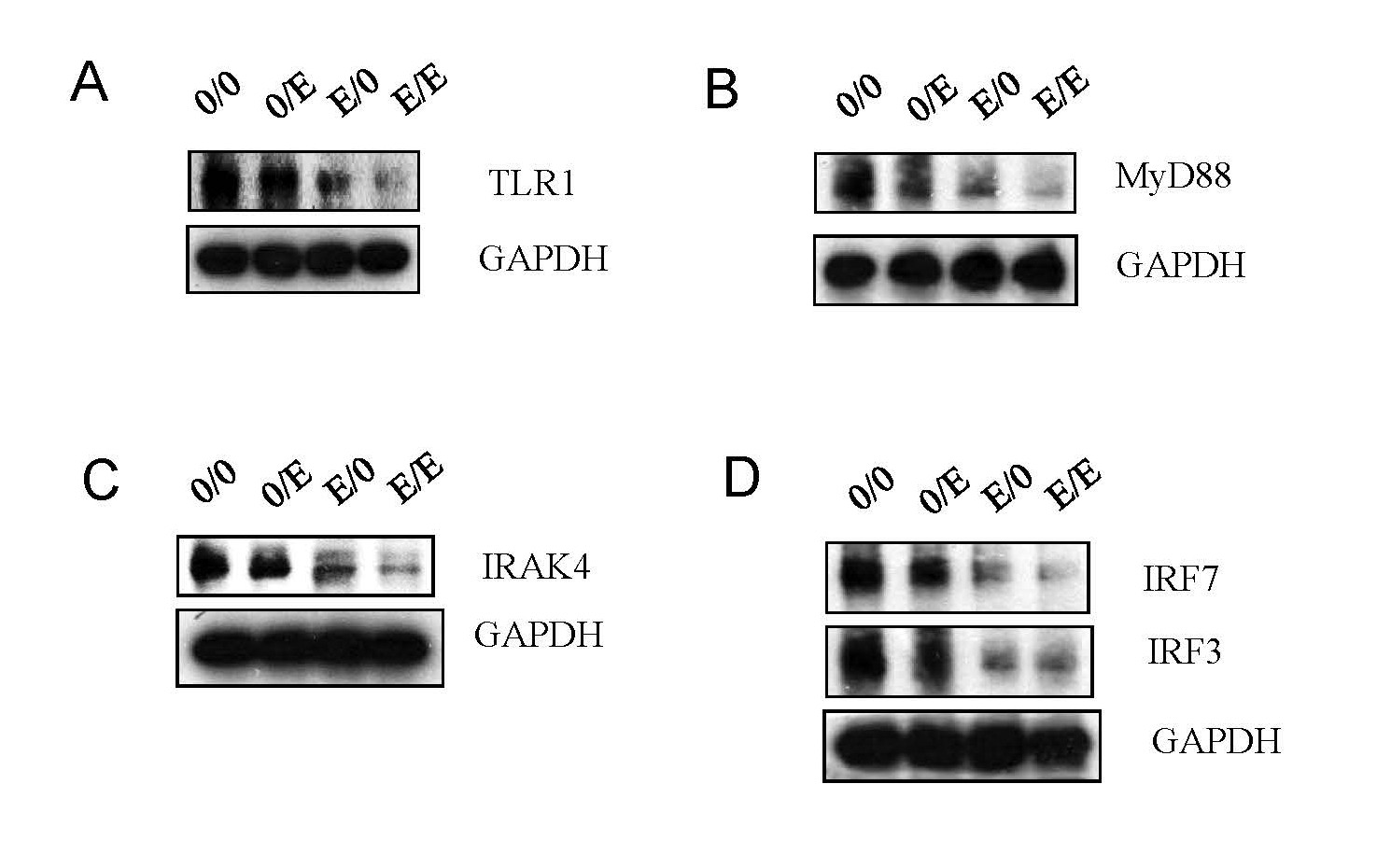
Figure 5. Emtricitabine pre-treatment blocked HIV-1 replication via TLR pathways. Jurkat cells were treated with 10 nM of emtricitabine or/and infected with HIV-1, as described in the Materials and Methods section and visually depicted in the Supplementary Figure 1. Total cell lysates were subjected to Western blot analysis to detect (A) TLR1; (B) MyD88; (C) IRAK4; and (D) IRF7 and IRF3.
FTCpt leads to the downregulation of Jak-Stat pathways
The Jak-Stat pathway, integral for immune responses [22-26], was impacted by emtricitabine pre-treatment. Downregulation of Jak1, Jak2, Stat5, Stat3, and Stat1 was observed, leading to reduced viral RNA production. Concomitantly, TF expression was upregulated. Emtricitabine post-treatment continued to suppress these Jak-Stat components, emphasizing their crucial role in the pathway (Figures 6A–6C), suggesting that emtricitabine pre-treatment inhibits Jak-Stat pathways, which are exploited by HIV-1 to evade host immune responses. The downregulation of key components suggests a potential disruption of interferon-stimulated genes (ISGs) responsible for antiviral responses.
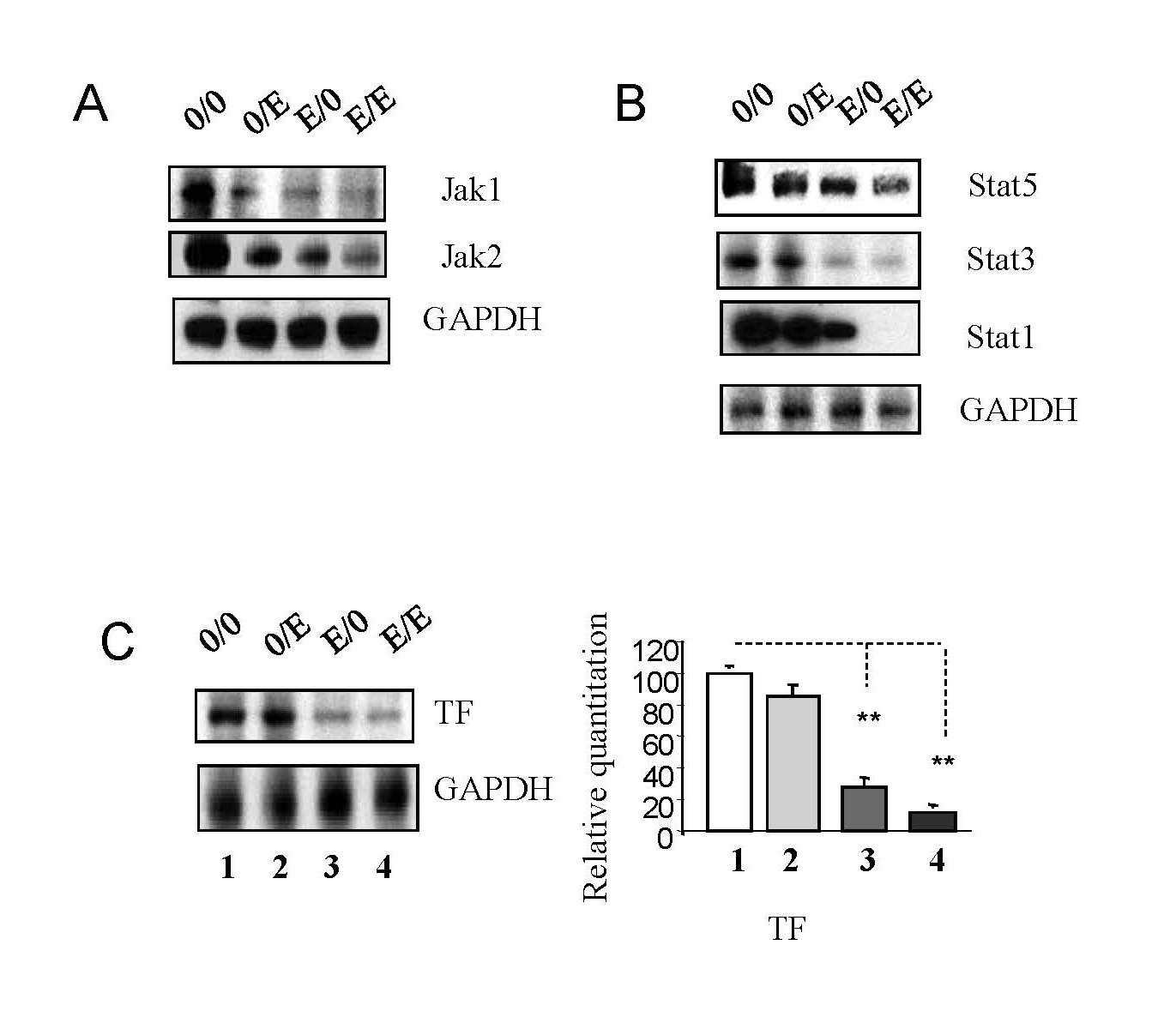
Figure 6. Emtricitabine pre-treatment decreased HIV-1 replication via Jak-Stat pathways. Jurkat cells were treated with 10 nM of emtricitabine or/and infected with HIV-1, as described in the Materials and Methods section and visually depicted in the Supplementary Figure 1. Total cell lysates were subjected to Western blot analysis to detect (A) Jak1 and Jak2; (B) Stat5, Stat3 and Stat1; and (C) TF.
Discussion and Conclusion
The establishment of a latent HIV-1 reservoir in CD4+ T cells remains a formidable challenge in achieving a cure for HIV-1 infection [27,28]. While ART effectively suppresses active viral replication in the bloodstream, it fails to eliminate latent reservoirs [29,30]. Emtricitabine, a key component of PrEP, provides a critical preventive measure but does not address the existing latent reservoirs [29,31]. The challenge lies in the persistence of latent HIV-1, necessitating ongoing research into therapeutic strategies that target these reservoirs.
PrEP has proven highly effective in preventing HIV transmission when taken consistently [10,11]. However, a notable challenge is the discontinuation of PrEP, with many individuals ceasing treatment within the first 3–6 months [10]. The study's findings underscore the importance of both pre- and post-treatment with emtricitabine for inhibiting HIV-1 replication and reducing viral loads. Stopping emtricitabine treatment may result in a resurgence of HIV-1 replication, emphasizing the need for sustained PrEP adherence [10].
HIV-1 replication involves intricate interactions with various cellular signaling pathways, highlighting the virus's adaptability and evasion mechanisms. The study explores the impact of emtricitabine on key pathways, including NF-κB [32], MAPK [8,9,33], TCR/PI3K-Akt [6,7], Jak-Stat [22-26], and TLR [20,21,34,35]. The data suggest that emtricitabine treatments significantly inhibit the activation of these pathways, crucial for HIV-1 replication and latency. This multi-pronged inhibition supports emtricitabine's potential as a broad-spectrum anti-HIV agent.
Previous research has implicated apoptotic signaling pathways in influencing HIV-1 replication [36] and latency [37]. Proapoptotic components have been associated with promoting HIV-1 replication [36,38], while antiapoptotic molecules exhibit the opposite effect [36,37]. The study proposes further investigation into how emtricitabine impacts apoptotic pathways induced by HIV-1 infection and replication. Understanding this interplay could provide insights into the regulation of HIV-1 replication and latency.
Emtricitabine pre-treatment exhibits a downregulation of key components involved in transcriptional machinery, such as Brd4, CDK9, Cyclin T1, and p-RPb-CTD (Figure 7). This suggests a potential mechanism for inhibiting the synthesis of full-length HIV-1 mRNA and maintaining the virus in a latent state. The downregulation of PCAF further supports the inactivation of HIV-1 replication through chromatin acetylation. Emtricitabine pre-treatment shows a significant downregulation of Jak-Stat pathway components, including Jak1, Jak2, Stat5, Stat3, and Stat1 (Figure 7). These components play a crucial role in immune responses, and their inhibition suggests a potential disruption of interferon-stimulated genes (ISGs) responsible for antiviral responses. Inhibition of the Jak-Stat pathway is a vital aspect of HIV-1 pathogenesis. The Toll-like receptor (TLR) pathway, essential for the initial recognition and response to HIV-1 infection, is inhibited by emtricitabine pre-treatment. Prolonged TLR activation has been linked to chronic immune activation and inflammation in HIV-1 infection. Emtricitabine's impact on the TLR pathway underscores its potential in modulating the immune response to the virus (Figure 7). Further studies are needed to address the role of phosphorylated STAT, which is commonly used as an indicator of protein activation, as well as the phosphorylation of key molecules in the PI3K/Akt and p38 MAPK pathways. Additionally, investigating the effects of inhibiting or knocking down proteins involved in TCR-related, PI3K-, and MAPK-mediated pathways may provide insights into mechanisms underlying viral load reactivation. The data obtained in this study were derived solely from Jurkat cells; additional cell models may be needed to better demonstrate the clinical relevance and significance of emtricitabine pre-treatment.
In conclusion, the study indicates that emtricitabine pre-treatment exhibits a multifaceted impact on HIV-1 replication (Figure 7). They hinder Tat-mediated recruitment, downregulate key transcriptional machinery, block TLR pathways crucial for immune response, and inhibit Jak-Stat pathways integral to antiviral defense. These findings shed light on the diverse mechanisms through which emtricitabine contributes to the inhibition of HIV-1 replication and highlights the importance of sustained treatment adherence. The multifaceted impact on various cellular pathways suggests emtricitabine's potential as a versatile therapeutic agent. Understanding these molecular interactions opens avenues for developing targeted strategies to address the persistent challenges posed by HIV-1 replication and latency. Further research is essential to unravel the complexities of emtricitabine's interactions with apoptotic pathways and its long-term impact on the HIV-1 life cycle.
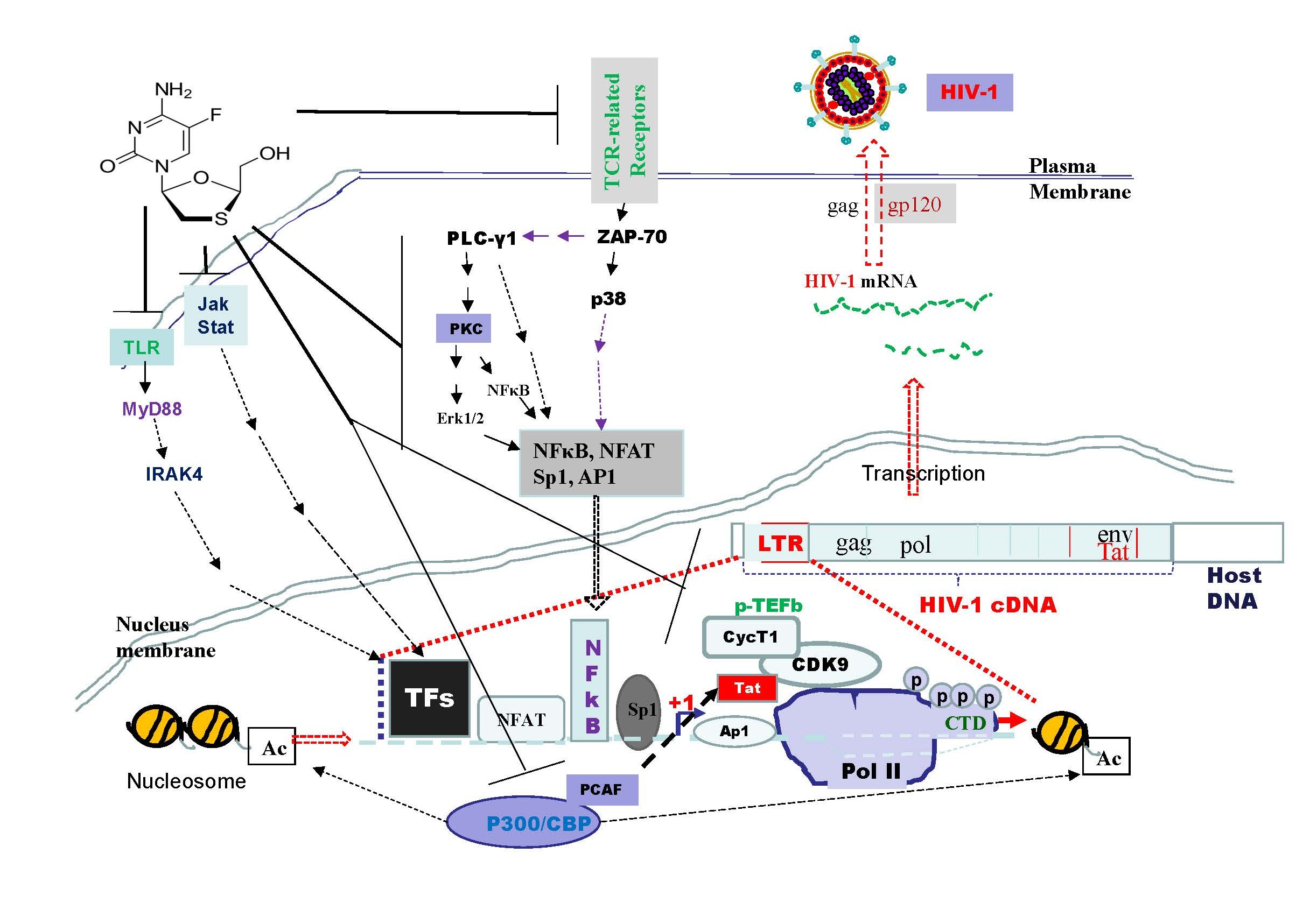
Figure 7. The model of emtricitabine pre-treatment on inhibition of HIV-1 infection in Jurkat cells. This inactivation of HIV-1 replication occurs through the downregulation of various factors such as NF-κB, p65, Ap-1, Sp-1, and NFAT in TCR/PI3K-Akt-related pathways. Additionally, decrease of STAT1, STAT3, and STAT5 in Jak/Stat pathways, as well as IRF3 and IRF7 in TLR-related pathways, contributes to this process. Furthermore, the inactivation of MAPK, ERK, and p38 activities is implicated. SARS-CoV-2 infection is also linked to the potential inhibition of full-length HIV-1 mRNA synthesis. This is achieved by decreasing p-TEFb signaling pathways and downregulating Brd4, CDK9, Cyclin T1, and p-RPb-CTD. Notably, the emtricitabine downregulates PCAF, which functions as histone acetyl transferases (HATs) for chromatin acetylation. This, in turn, inhibits HIV-1 replication.
Abbreviations
AIDS: Acquired Immunodeficiency Syndrome; Akt: Protein kinase B; Ap-1: Activator protein 1; ART: Antiretroviral Therapy; Brd4: Bromodomain containing protein 4; CD: Cluster of Differentiation; CDK9: Cyclin-Dependent Kinase 9; CTD: Carboxyl-Terminal Domain; Erk: Extracellular signal-regulated kinase; HAT: Histone Acetyl Transferase; HIV-1: Human Immunodeficiency Virus type-1; IRAK-4: IL-1 Receptor-Associated Kinase-4; IRF: Interferon Regulatory Factor; Jak: Janus kinase; LTR: Long Terminal Repeat; MAPK: Mitogen-Activated Protein Kinase; MyD88: Myeloid Differentiation primary response 88; NFAT: Nuclear Factor of Activated T cells; NF-κB: Nuclear Factor κB; p38: p38 Mitogen-Activated Protein Kinase; PCAF: P300/CBP-Associated Factor; PI3K: Phosphoinositide 3-kinase; PKC: Protein Kinase C; PLCγ-1: Phospholipase C gamma1; pol II: RNA polymerase II; PrEP: Pre-Exposure Prophylaxis; P-RPb-CTD: Phospho-Rpb1 CTD; p-TEFb: positive Transcription Elongation Factor b; RPb: the largest subunit of RNA polymerase II; Sp-1: Specificity protein 1; Stat: Signal transducer and activator of transcription; TAR: Transactivation Response Element; Tat: Trans-activator of transcription; TCR: T cell Receptor; TF: Transcription Factor; TLR: Toll-Like Receptor; Zap-70: Zeta-chain-associated protein kinase 70
Author Contributions
XW and IH conceived the project. XW and JZ performed the experiments. XW formatted datasets and analyzed data. XW and IH participated in manuscript writing, engaged in data interpretation, and contributed to the intellectual content of this work.
Acknowledgments
The authors wish to acknowledge Dr. Santanu Biswas and Dr. Mohan Haleyurgirisetty, for their critical review of this manuscript. The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any Agency determination or policy.
Competing Interests
The authors declare no competing interests and no fundings.
Data Availability
The datasets of this research are available from the corresponding author on request.
Consent for Publication and Ethical Approval
Not applicable.
Funding
Not applicable.
References
2. SeyedAlinaghi S, Afsahi AM, Moradi A, Parmoon Z, Habibi P, Mirzapour P, et al. Current ART, determinants for virologic failure and implications for HIV drug resistance: an umbrella review. AIDS Res Ther. 2023 Oct 27;20(1):74.
3. Mbonye U, Karn J. Transcriptional control of HIV latency: cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology. 2014 Apr;454-455:328-39.
4. Bermejo M, López-Huertas MR, Hedgpeth J, Mateos E, Rodríguez-Mora S, Maleno MJ, et al. Analysis of protein kinase C theta inhibitors for the control of HIV-1 replication in human CD4+ T cells reveals an effect on retrotranscription in addition to viral transcription. Biochem Pharmacol. 2015 Apr 15;94(4):241-56.
5. Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591-619.
6. Somech R. T-cell receptor excision circles in primary immunodeficiencies and other T-cell immune disorders. Curr Opin Allergy Clin Immunol. 2011 Dec;11(6):517-24.
7. asquereau S, Herbein G. CounterAKTing HIV: Toward a "Block and Clear" Strategy? Front Cell Infect Microbiol. 2022 Feb 4;12:827717.
8. Wang X, Zhao J, Ragupathy V, Hewlett I. The effects of MAPK p38α on AZT resistance against reactivating HIV-1 replication in ACH2 cells. Mol Cell Biochem. 2019 Dec;462(1-2):41-50.
9. Furler RL, Uittenbogaart CH. Signaling through the P38 and ERK pathways: a common link between HIV replication and the immune response. Immunol Res. 2010 Dec;48(1-3):99-109.
10. Chou R, Spencer H, Bougatsos C, Blazina I, Ahmed A, Selph S. Pre-Exposure Prophylaxis for the Prevention of HIV Infection: A Systematic Review for the U.S. Preventive Services Task Force [Internet]. Rockville (MD): Agency for Healthcare Research and Quality (US); 2023; Report No.: 22-05300-EF-1.
11. Piliero PJ. Pharmacokinetic properties of nucleoside/nucleotide reverse transcriptase inhibitors. J Acquir Immune Defic Syndr. 2004 Sep 1;37 Suppl 1:S2-S12.
12. Wang X, Sun B, Mbondji C, Biswas S, Zhao J, Hewlett I. Differences in Activation of HIV-1 Replication by Superinfection With HIV-1 and HIV-2 in U1 Cells. J Cell Physiol. 2017 Jul;232(7):1746-53.
13. Wang X, Tan J, Zhao J, Ragupathy V, Haleyurgirisetty M, Hewlett I. Some findings of FADD knockdown in inhibition of HIV-1 replication in Jurkat cells and PBMCs. Mol Cell Biochem. 2014 Aug;393(1-2):181-90.
14. Institute for Quality and Efficiency in Health Care. Fixed-dose combination of bictegravir/emtricitabine/tenofovir alafenamide (Biktarvy) for the treatment of HIV: Overview. 2018 In InformedHealth.org.
15. Gotora PT, van der Sluis R, Williams ME. HIV-1 Tat amino acid residues that influence Tat-TAR binding affinity: a scoping review. BMC Infect Dis. 2023 Mar 17;23(1):164.
16. Pumfery A, Deng L, Maddukuri A, de la Fuente C, Li H, Wade JD, et al. Chromatin remodeling and modification during HIV-1 Tat-activated transcription. Curr HIV Res. 2003 Jul;1(3):343-62.
17. Liu RD, Wu J, Shao R, Xue YH. Mechanism and factors that control HIV-1 transcription and latency activation. J Zhejiang Univ Sci B. 2014 May;15(5):455-65.
18. Fujinaga K. P-TEFb as A Promising Therapeutic Target. Molecules. 2020 Feb 14;25(4):838.
19. Rice AP. Roles of CDKs in RNA polymerase II transcription of the HIV-1 genome. Transcription. 2019 Apr;10(2):111-7.
20. Martinsen JT, Gunst JD, Højen JF, Tolstrup M, Søgaard OS. The Use of Toll-Like Receptor Agonists in HIV-1 Cure Strategies. Front Immunol. 2020 Jun 11;11:1112.
21. Kayesh ME, Kohara M, Tsukiyama-Kohara K. Toll-like receptor response to human immunodeficiency virus type 1 or co-infection with hepatitis B or C virus: an overview. Int J Mol Sci. 2023 Jun 1;24(11):9624.
22. Mahjoor M, Mahmoudvand G, Farokhi S, Shadab A, Kashfi M, Afkhami H. Double-edged sword of JAK/STAT signaling pathway in viral infections: novel insights into virotherapy. Cell Commun Signal. 2023 Oct 2;21(1):272.
23. Rhodes DA, de Bono B, Trowsdale J. Relationship between SPRY and B30.2 protein domains. Evolution of a component of immune defence? Immunology. 2005; 116(4): 411-7.
24. Venkatachari NJ, Zerbato JM, Jain S, Mancini AE, Chattopadhyay A, Sluis-Cremer N, et al. Temporal transcriptional response to latency reversing agents identifies specific factors regulating HIV-1 viral transcriptional switch. Retrovirology. 2015 Oct 6;12:85.
25. Gavegnano C, Detorio M, Montero C, Bosque A, Planelles V, Schinazi RF. Ruxolitinib and tofacitinib are potent and selective inhibitors of HIV-1 replication and virus reactivation in vitro. Antimicrob Agents Chemother. 2014;58(4):1977-86.
26. Chung H, Green PHR, Wang TC, Kong XF. Interferon-Driven Immune Dysregulation in Down Syndrome: A Review of the Evidence. J Inflamm Res. 2021 Oct 7;14:5187-200.
27. Mori L, Valente ST. Cure and Long-Term Remission Strategies. Methods Mol Biol. 2022;2407:391-428.
28. Pasternak AO, Berkhout B. HIV persistence: silence or resistance? Curr Opin Virol. 2023 Apr;59:101301.
29. Sibiude J, Le Chenadec J, Mandelbrot L, Hoctin A, Dollfus C, Faye A, et al. Update of Perinatal Human Immunodeficiency Virus Type 1 Transmission in France: Zero Transmission for 5482 Mothers on Continuous Antiretroviral Therapy From Conception and With Undetectable Viral Load at Delivery. Clin Infect Dis. 2023 Feb 8;76(3):e590-8.
30. Alessandri-Gradt E, Moisan A, Plantier JC. HIV-1 Non-Group M Strains and ART. Viruses. 2023 Mar 17;15(3):780.
31. D'Orso I, Forst CV. Mathematical Models of HIV-1 Dynamics, Transcription, and Latency. Viruses. 2023 Oct 19;15(10):2119.
32. Deng L, Zeng Q, Wang M, Cheng A, Jia R, Chen S, et al. Suppression of NF-κB Activity: A Viral Immune Evasion Mechanism. Viruses. 2018 Aug 4;10(8):409.
33. Liu X, Shah A, Gangwani MR, Silverstein PS, Fu M, Kumar A. HIV-1 Nef induces CCL5 production in astrocytes through p38-MAPK and PI3K/Akt pathway and utilizes NF-kB, CEBP and AP-1 transcription factors. Sci Rep. 2014 Mar 24;4:4450.
34. Hokello J, Lakhikumar Sharma A, Tyagi M. AP-1 and NF-κB synergize to transcriptionally activate latent HIV upon T-cell receptor activation. FEBS Lett. 2021 Mar;595(5):577-94.
35. Barclay RA, Mensah GA, Cowen M, DeMarino C, Kim Y, Pinto DO, et al. Extracellular Vesicle Activation of Latent HIV-1 Is Driven by EV-Associated c-Src and Cellular SRC-1 via the PI3K/AKT/mTOR Pathway. Viruses. 2020 Jun 19;12(6):665.
36. Wang X, Ragupathy V, Zhao J, Hewlett I. Molecules from apoptotic pathways modulate HIV-1 replication in Jurkat cells. Biochem Biophys Res Commun. 2011 Oct 14;414(1):20-4.
37. Wang X, Zhao J, Biswas S, Devadas K, Hewlett I. Components of apoptotic pathways modulate HIV-1 latency in Jurkat cells. Microbes Infect. 2022 Apr-May;24(3):104912.
38. Wang X, Viswanath R, Zhao J, Tang S, Hewlett I. Changes in the level of apoptosis-related proteins in Jurkat cells infected with HIV-1 versus HIV-2. Mol Cell Biochem. 2010 Apr;337(1-2):175-83.