Abstract
Glucocorticoids are steroid hormones produced by the adrenal cortex in a circadian manner and they participate in many physiological and pathological processes. Synthetic glucocorticoids have been universally applied to treat inflammatory diseases and immune disorders. Due to their angiostatic property, glucocorticoids are often added to regimens for cancer treatment. In the current review, we summarize how glucocorticoids influence angiogenesis in common solid tumors based on literature from the last ten years. Usage of glucocorticoids can be a double-edged sword in the treatment of some malignancies. There are still unanswered questions about the role of glucocorticoids in the treatment regimens of some common cancers. Therefore, we suggest prudent and restricted administration of glucocorticoids to treat solid tumors.
Keywords
Glucocorticoids, Angiogenesis, Malignancy
Glucocorticoids and the Glucocorticoid Receptor
Glucocorticoids (GCs) are defined by their role in maintaining glucose homeostasis and natural GCs are a class of corticosteroids secreted by the adrenal cortex [1]. Cortisol is the most important natural GC in humans. Cellular cortisol levels are regulated by the tissue-specific metabolic enzymes 11β-hydroxysteroid dehydrogenase 1 and 2 (11β-HSD 1 and 2); 11β-HSD 1 converts inactive cortisone to active cortisol, while 11β-HSD 2 has the opposite function [2]. The relative activity of these enzymes is responsible for maintaining the balance of cortisol in vivo. The release of cortisol into the circulation is involved in a variety of systemic processes such as immune responses, metabolism, cell growth, development, and reproduction [3]. Additionally, due to the multi-functional features of GCs, more and more synthetic GCs, such as hydrocortisone, dexamethasone (DEX), prednisone (PRED), triamcinolone acetonide (TA) and budesonide (BUD) are being widely-prescribed in clinical settings [4,5].
GCs trigger gene transcription by interacting with the glucocorticoid receptor (GR). GR is encoded by the gene NR3C1 and acts as a ligand-inducible transcription factor [6]. By specific alternative splicing, GR has several common isoforms, including GRα and GRβ. GRα is the classical GR isoform which has strong binding affinity for GCs, while the GRβ splice variant does not bind GCs, instead functioning as a natural inhibitor of the GRα isoform on many GC-responsive target genes [7]. Increased expression of GRβ has been associated with GC resistance, which may be due to competition for transcriptional co-regulators, or the formation of inactive GRα/GRβ heterodimers [2].
GC-GR signaling functions in two ways: via genomic effects and via non-genomic effects [8]. The former is the canonical manner which depends on GR-mediated transcription and protein synthesis. Once binding with GCs, GR rapidly translocates to the nucleus and subsequently regulates the transcription of its target genes through genomic mechanisms [9]. Unlike the genomic effect, which usually takes place in hours, the non-genomic effect of GC-GR signaling can arise within minutes because such an action is initiated at the cell surface via either membrane binding [10] or cytoplastic GR [11], rather than requiring GR to translocate to the nucleus. Due to different mechanisms between genomic and non-genomic effects of GC-GR signaling, GCs also exert distinct actions in different diseases.
Mechanisms of GC Regulation of Angiogenesis
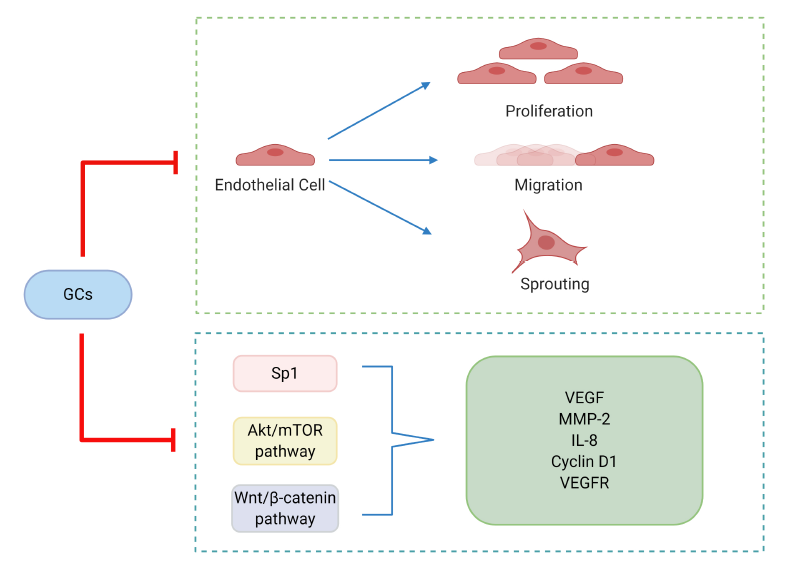
GCs are angiostatic and are used to treat angiogenesisrelated diseases, including diabetic retinopathy and solid tumors. Several studies illustrate how GCs regulate angiogenesis. As shown by Shikatani et al., corticosterone decreased the number of capillaries in rat skeletal muscle and inhibited proliferation, migration, and sprouting of skeletal muscle microvascular endothelial cells in vitro [12]. After treatment with corticosterone, endothelial cells showed diminished VEGF mRNA levels, as well as reduced production and activation of MMP-2, correlating with inhibition of cell sprouting within a 3D collagen matrix. The expression of Sp1, a transcriptional regulator of both VEGF and MMP-2, was also demonstrated to be down-regulated by corticosterone. In vitro, TA treatment decreased the protein expression of the cellular and soluble forms of VEGF and VEGFR-1 as well as vascular network forming capacity through the Akt/mTOR pathway in a time- and dose-dependent manner [13].
The Wnt/β-catenin signaling pathway also appears to be critical in vascular endothelial cells and acts through a variety of regulators, including Bach1 [14], Rspo1 [15], Endostar [16], and ERG [17]. Once Wnt/β-catenin signaling is triggered, β-catenin binds to TCF in the nucleus, and alters the expression of key drivers and regulators of angiogenesis, such as VEGF, IL-8, Cyclin D1 and MMP-2 [18]. Using a ChIP-seq approach as well as a validate mouse model, we have recently shown that loss of endothelial GR results in the upregulation of Wnt signaling both in vitro and in vivo [19]. Therefore, we hypothesize that steroid microenvironments in endothelial cell networks have important implications for regulation of angiogenesis. The regulatory effects of GCs on angiogenesis are shown in Figure 1.
In general, GCs can regulate angiogenesis via two main approaches: (i) suppression of proliferation, migration and sprouting in endothelial cells and (ii) reduction of the secretion or expression of key cytokines and/or proteins responsible for the upregulation of angiogenesis.
Role of GCs in Cancer Treatment
Synthetic GCs have been globally applied for the treatment of inflammatory and immune disorders, including rheumatoid arthritis, multiple sclerosis, inflammatory bowel disease, and nephrotic syndrome [20]. In addition, owing to their ability to induce apoptosis in hematological cells, GCs are used as chemotherapeutic agents for the treatment of acute lymphoblastic leukemia (ALL), chronic lymphoblastic leukemia (CLL), multiple myeloma (MM), Hodgkin’s lymphoma (HL) and non- Hodgkin’s lymphoma (NHL) [21-25].
For non-hematologic malignancies, GCs can have either adjuvant or curative effects, depending on the subtype of tumor as well as the specific treatment protocols. For instance, due to their anti-emetic and anti-edemic properties, GC administration is nearly always added to surgery, radiotherapy or chemotherapy, where they can relieve symptoms of the primary disease, alleviate side effects of chemotherapy, and protect healthy tissues from cytotoxic effects induced by chemotherapeutic treatment [26-28].
In breast cancer, data from an animal model [29] demonstrated the treatment with TA decreased capsular thickness of the tumor, mild mononuclear inflammation, and negative or minimal angiogenesis in rabbits. However, according to Flaherty’s study, both in 66CL4 breast cancer cells and the mouse breast cancer model, GCs can induce DNA damage through an inducible nitric oxide synthase (iNOS)- mediated pathway by increasing levels of nitric oxide (NO); increased NO further stimulated by GC signaling may serve to promote angiogenesis through VEGF in a chronic stress model [30].
In prostate cancer, Yano et al. revealed that GCs acted directly through GR and suppressed two major angiogenic factors, VEGF and IL-8, in the androgen-independent prostate cancer cell line DU145. Additionally, in a xenograft model, except for intratumor VEGF and IL-8 gene expression, DEX treatment also inhibited angiogenesis and in vivo tumor growth [31]. Nevertheless, evidence exists that the GC signaling pathway can increase the diameter of blood vessels and vessel area in tumor tissues from prostate cancer patients [32].
In bladder cancer, Ishiguro et al. [33] showed that both DEX and PRED could repress the expression of MMP- 9, VEGF, and IL-6 in UMUC3 and TCC-SUP human urothelial carcinoma cell lines. However, another study evaluated the effects of DEX on cell proliferation, apoptosis, and invasion in bladder cancer cells lines and found that, although DEX impeded cell invasion and the expression of angiogenesis-related genes (MMP-2/MMP- 9, IL-6, and VEGF), as well as induced mesenchymal-toepithelial transition, it also correlated positively with cell proliferation in mouse xenograft models and resulted in a significant reduction in the curative effects of cisplatin [34].
In glioblastoma multiforme (GBM), DEX treatment didn’t result in any changes either in total vessel area or average vessel size compared to vehicle treatment in a mouse model. Furthermore, clinical data implied that GCs might decrease the effectiveness of radiotherapy and chemotherapy as well as reduce overall survival in GBM patients [35]. According to Llaguno-Munive’s study [36], mifepristone (Mife), which is considered an antiglucocorticoid, was used in combination with chemoradiotherapy (Rad) and Temozolomide (Tmz) to treat GBM in mice. After 25 days, the tumor volume of the Rad + Tmz + Mife group was significantly less than that of the Rad + Tmz group. Furthermore, the expression of VEGF also decreased in the Rad + Tmz + Mife group. Several other clinical studies also showed that a decrease in GC use could improve the prognosis of GBM [37,38].
In melanoma, a new class of cationic lipid–DEX conjugate containing a C-8 carbon chain analogue (DX8), was used to study the efficiency of GCs on tumorbearing mice. After calculating the area stained by VEGFR2 in endothelial cells, DX8 decreased the level of VEGFR2 in tumor-endothelial cells, implying DX8’s antiangiogenic role in melanoma [39]. As demonstrated in Licarete’s study [40], the administration of prednisolone disodium phosphate (PLP) could improve doxorubicin cytotoxicity on B16.F10 murine melanoma cells in vitro via the inhibition of the proangiogenic function of tumorassociated macrophages (TAMs). Another study [41] revealed that HYC16, a kind of cationic lipid modification of hydrocortisone, exhibited significantly less VEGFR2 expression and lower density of vascular endothelial cells in mice, indicating HYC16 had an evident antiangiogenic effect and substantiated its ability to inhibit tumor growth.
In HCT116 and HT29 colon cancer cell lines, DEX treatment inhibited HIF-1α protein levels and its downstream gene, VEGF mRNA levels. Also, the presence of DEX suppressed the mRNA levels of hypoxia-induced Snail, Slug, and Twist as well as hypoxia-induced integrin αVβ6 protein levels, which is a well-known EMT marker for colon cancer cells [42]. Based on Patras’s study [43], prednisolone-loaded long-circulating liposomes (LCLPLP) + LCL-5-FU combination therapy resulted in lower expression of M-CSF, MCP-1, eotaxin, leptin, G-CSF, IGF-II, IL-1α, IL-1β, IL-9, IL-12p40, FasL, bFGF, and VEGF in C26 colon carcinoma tissue, which implies antiinflammatory and anti-angiogenic effects of LCL-PLP.
In a rat liver cancer model, compared to a glucuronolactone alone group, tumor nodule number and micro vessel density in the glucuronolactone + hydrocortisone group were significantly lower at week 12. Additionally, significantly decreased levels of macrophages, TNF-α, p-p38, NF-κB, IL-10, HGF, TGF-β1, and VEGF were observed in the paraneoplastic tissue of the glucuronolactone + hydrocortisone group when compared with the glucuronolactone group. The results suggest that hydrocortisone treatment reduces macrophage polarization, inflammatory and antiinflammatory cytokines levels, and angiogenesis in paraneoplastic tissue [44]. Similar results were also obtained in a mice model as the tumor weight in the DEX treatment group was observed to be significantly lower than that in the control group. Both tumor blood vessel density and total blood vessel length in the DEX group were smaller than those in the control group. These results indicate that DEX has an inhibitive effect on tumor growth and angiogenesis in murine liver cancer in situ [45].
As reported in Geng’s study [46], Lewis lung carcinoma cells were inoculated in C57BL/6 mice, and the mice were randomly divided into 3 groups: a control group, a cisplatin group, and a DEX group. The results demonstrated that tumor growth was suppressed in the both cisplatin group and the DEX groups. In addition, tumor weights decreased in the cisplatin and DEX groups compared to the control group. The expression of HIF- 1α and VEGF and the density of micro vessels were also significantly lower in the cisplatin and DEX groups than in the control group. However, these changes were not significantly different between the cisplatin group and DEX group, indicating DEX could effectively inhibit the growth and angiogenesis of Lewis lung carcinoma to the same extent as cisplatin, by suppressing the expression of HIF-1α and VEGF. Sun’s study also achieved a similar conclusion [47].
Overall, the anti-angiogenic mechanisms of GCs in cancer treatment can be summarized as follows: either (a) direct effects on tumor-derived vasculature and other cellular populations from the tumor microenvironment and (b) indirect effects via affecting cancer cell-derived factors [4]. In different subtypes of cancer, GCs regulate angiogenesis in diverse manners, including upregulation and downregulation (Table 1).
| Cancer | GC subtype | Species | Effect on angiogenesis |
Other effects | Ref |
|---|---|---|---|---|---|
| Breast cancer | TA | Rabbit | Down-regulation | - | [29] |
| Breast cancer | Cortisol | Mouse | Up-regulation | - | [30] |
| Prostate cancer | DEX | Mouse | Down-regulation | - | [31] |
| Prostate cancer | GC signaling pathway | Human | Up-regulation | - | [32] |
| Bladder cancer | DEX, PRED | Cell line | Down-regulation | - | [33] |
| Bladder cancer | DEX | Mouse | Down-regulation | Decrease the effectiveness of chemotherapy |
[34] |
| GBM | DEX | Human | None | Decrease the effectiveness of radiotherapy and chemotherapy | [35] |
| GBM | Mife (Antagonist of GC) | Mouse | Down-regulation | - | [36] |
| Melanoma | DX8 | Mouse | Down-regulation | - | [39] |
| Melanoma | PLP | Cell line | Down-regulation | - | [40] |
| Melanoma | HYC16 | Mouse | Down-regulation | - | [41] |
| Colon cancer | DEX | Cell line | Down-regulation | - | [42] |
| Colon cancer | LCL-PLP | Mouse | Down-regulation | - | [43] |
| Liver cancer | Hydrocortisone | Rat | Down-regulation | - | [44] |
| Liver cancer | DEX | Mouse | Down-regulation | - | [45] |
| Lung cancer | DEX | Mouse | Down-regulation | - | [46] |
| Lung cancer | DEX | Mouse | Down-regulation | - | [47] |
Table 1: The effects of GCs on angiogenesis in particular solid tumors.
Outstanding Questions
Though the role of GCs in the anti-angiogenic treatment of particular types of solid tumors is clear, there are other tumors for which GC treatment remains unclear. For example, in gastrointestinal cancer Busada et al. removed circulating GCs in mice by adrenalectomy, and showed the rapid onset of spontaneous gastric inflammation, oxyntic atrophy, and spasmolytic polypeptide-expressing metaplasia (SPEM), a putative precursor of gastric cancer [48]. Therefore, the authors hypothesized that endogenous glucocorticoid signaling was essential in preventing spontaneous gastric inflammation and metaplasia as well as gastric cancer development.
In renal cancer, an in vitro study investigated the relationship between Mife and apoptosis. Human renal carcinoma cell (RCC) lines, Caki, A498, ACHN, HT29, and SK-Hep1 were treated with Mife and the results revealed Mife enhanced the sensitivity of RCCs to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)- induced apoptosis through induction of DR5 expression and reduction of Bcl-2 and c-FLIP(L) expression [49]. However, the effect of Mife on TRAIL sensitization was independent of GR signaling.
In ovarian cancer, a study in 2006 demonstrated premedication with DEX prior to cisplatin or gemcitabine abrogated the growth-inhibitory or apoptotic response of the chemotherapeutic agents in the ovarian carcinoma cell lines, SKOV3, OAW-42, OVM, and M130 [50]. Furthermore, xenograft tumors in mice treated with DEX and cisplatin grew as fast in vivo as untreated controls, which indicated that the presence of GCs reduced the efficiency of chemotherapy. Additionally, another in vitro study [51] demonstrated that cytotoxic cisplatin and/or paclitaxel treatment decreased cellular attachment by 51% and resulted in significant cell death as compared to controls. But when simultaneously administered with cisplatin and/or paclitaxel treatment, DEX increased cell survival and adhesion in parallel and in a dose-dependent manner, implying DEX completely blocked apoptosis induction by cytotoxic treatment. Therefore, the use of GCs in the treatment of ovarian cancer is currently discouraged.
Conclusions
Due to the anti-angiogenic feature of GCs, they are now regarded as an effective treatment for solid tumors. However, our review of the literature from the last ten years, reveals that the administration of GCs can be a double-edged sword in cancer therapy. Especially for breast cancer and prostate cancer, the introduction of GCs might promote angiogenesis under certain conditions. For the treatment of bladder cancer and GBM, the administration of GCs might decrease the effectiveness of radiotherapy and chemotherapy. But in melanoma, colon cancer and liver cancer, the angiostatic effect of GCs seems evident and straightforward. These discrepancies highlight the pleiotropic effects of GCs in different tumor environments. Therefore, considering the established adverse effect profile of GCs, we strongly suggest a prudent and individualized use of GCs in the treatment of solid tumors.
Abbreviations
ALL: Acute Lymphoblastic Leukemia; BUD: Budesonide; CLL: Chronic Lymphoblastic Leukemia; DEX: Dexamethasone; GBM: Glioblastoma Multiforme; GC: Glucocorticoid; GR: Glucocorticoid Receptor; HL: Hodgkin’s Lymphoma; iNOS: inducible Nitric Oxide Synthase; LCL: Long-Circulating Liposome; Mife: Mifepristone; MM: Multiple Myeloma; NHL: Non- Hodgkin’s Lymphoma; NO: Nitric Oxide; PLP: Pyridoxal Phosphate; PRED: Prednisone; Rad: Radiotherapy; RCC: Renal Carcinoma Cell; SPEM: Spasmolytic Polypeptide- Expressing Metaplasia; TA: Triamcinolone Acetonide; Tmz: Temozolomide; 11β-HSD 1, 2: 11β-Hydroxysteroid Dehydrogenase 1, 2
Funding
JEG is supported by the National Heart, Lung and Blood Institute (R01HL131952).
References
2. Liu B, Zhang TN, Knight JK, Goodwin JE. The Glucocorticoid Receptor in Cardiovascular Health and Disease. 2019 Oct 9;8(10):1227.
3. Cain DW, Cidlowski JA. Specificity and sensitivity of glucocorticoid signaling in health and disease. Best Practice & Research Clinical Endocrinology & Metabolism. 2015 Aug 1;29(4):545-56.
4. Martens B, Drebert Z. Glucocorticoid-mediated effects on angiogenesis in solid tumors. The Journal of Steroid Biochemistry and Molecular Biology. 2019 Apr 1;188:147-55.
5. Paragliola RM, Papi G, Pontecorvi A, Corsello SM. Treatment with synthetic glucocorticoids and the hypothalamus-pituitary-adrenal axis. International Journal of Molecular Sciences. 2017 Oct;18(10):2201.
6. Lavery DN, Mcewan IJ. Structure and function of steroid receptor AF1 transactivation domains: induction of active conformations. Biochemical Journal. 2005 Nov 1;391(3):449-64.
7. Kino T, Su YA, Chrousos GP. Human glucocorticoid receptor isoform β: recent understanding of its potential implications in physiology and pathophysiology. Cellular and Molecular Life Sciences. 2009 Nov 1;66(21):3435-48.
8. Stahn C, Buttgereit F. Genomic and nongenomic effects of glucocorticoids. Nature Clinical Practice Rheumatology. 2008 Oct;4(10):525-33.
9. Vandevyver S, Dejager L, Libert C. Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocrine Reviews. 2014 Aug 1;35(4):671-93.
10. Talabér G, Boldizsár F, Bartis D, Pálinkás L, Szabo M, Berta G, et al. Mitochondrial translocation of the glucocorticoid receptor in double-positive thymocytes correlates with their sensitivity to glucocorticoidinduced apoptosis. International Immunology. 2009 Nov 1;21(11):1269-76.
11. Panettieri RA, Schaafsma D, Amrani Y, Koziol- White C, Ostrom R, Tliba O. Non-genomic effects of glucocorticoids: an updated view. Trends in pharmacological sciences. 2019 Jan 1;40(1):38-49.
12. Shikatani EA, Trifonova A, Mandel ER, Liu ST, Roudier E, Krylova A, Szigiato A, Beaudry J, Riddell MC, Haas TL. Inhibition of proliferation, migration and proteolysis contribute to corticosterone-mediated inhibition of angiogenesis. PloS one. 2012;7(10):e46625.
13. Ozmen A, Unek G, Kipmen-Korgun D, Mendilcioglu I, Sanhal C, Sakıncı M, et al. Glucocorticoid effects on angiogenesis are associated with mTOR pathway activity. Biotechnic & Histochemistry. 2016 May 18;91(4):296- 306.
14. Jiang L, Yin M, Wei X, Liu J, Wang X, Niu C, et al. Bach1 represses Wnt/β-catenin signaling and angiogenesis. Circulation Research. 2015 Jul 31;117(4):364-75.
15. Gore AV, Swift MR, Cha YR, Lo B, McKinney MC, Li W, et al. Rspo1/Wnt signaling promotes angiogenesis via Vegfc/Vegfr3. Development. 2011 Nov 15;138(22):4875- 86.
16. Xu X, Mao W, Chen Q, Zhuang Q, Wang L, Dai J, et al. Endostar, a modified recombinant human endostatin, suppresses angiogenesis through inhibition of Wnt/β- catenin signaling pathway. PloS one. 2014;9(9):e107463.
17. Birdsey GM, Shah AV, Dufton N, Reynolds LE, Almagro LO, Yang Y, et al. The endothelial transcription factor ERG promotes vascular stability and growth through Wnt/β-catenin signaling. Developmental Cell. 2015 Jan 12;32(1):82-96.
18. Olsen JJ, Pohl SÖ, Deshmukh A, Visweswaran M, Ward NC, Arfuso F, Agostino M, Dharmarajan A. The role of Wnt signalling in angiogenesis. The Clinical Biochemist Reviews. 2017 Nov;38(3):131-142.
19. Zhou H, Mehta S, Srivastava SP, Grabinska K, Zhang X, Wong C, et al. Endothelial cell–glucocorticoid receptor interactions and regulation of Wnt signaling. JCI insight. 2020 Feb 13;5(3).
20. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. New England Journal of Medicine. 2005 Oct 20;353(16):1711-23.
21. Jing D, Bhadri VA, Beck D, Thoms JA, Yakob NA, Wong JW, et al. Opposing regulation of BIM and BCL2 controls glucocorticoid-induced apoptosis of pediatric acute lymphoblastic leukemia cells. Blood, The Journal of the American Society of Hematology. 2015 Jan 8;125(2):273-83.
22. Melarangi T, Zhuang J, Lin K, Rockliffe N, Bosanquet AG, Oates M, et al. Glucocorticoid resistance in chronic lymphocytic leukaemia is associated with a failure of upregulated Bim/Bcl-2 complexes to activate Bax and Bak. Cell Death & Disease. 2012 Aug;3(8):e372.
23. Burwick N, Sharma S. Glucocorticoids in multiple myeloma: past, present, and future. Annals of hematology. 2019 Jan 30;98(1):19-28.
24. Borchmann P, Goergen H, Kobe C, Lohri A, Greil R, Eichenauer DA, et al. PET-guided treatment in patients with advanced-stage Hodgkin’s lymphoma (HD18): final results of an open-label, international, randomised phase 3 trial by the German Hodgkin Study Group. The Lancet. 2017 Dec 23;390(10114):2790-802.
25. Adams HJ, Kwee TC. The Deauville criteria cannot differentiate between responding and non-responding non-Hodgkin lymphoma patients. Annals of Hematology. 2018 Apr 1;97(4):719-20.
26. Djedovic V, Lee YY, Kollara A, May T, Brown TJ. The Two Faces of Adjuvant Glucocorticoid Treatment in Ovarian Cancer. Hormones and Cancer. 2018 Apr 1;9(2):95-107.
27. Montgomery B, Cheng HH, Drechsler J, Mostaghel EA. Glucocorticoids and prostate cancer treatment: friend or foe?. Asian Journal of Andrology. 2014 May;16(3):354.
28. Maurice-Dror C, Perets R, Bar-Sela G. Glucocorticoids as an adjunct to oncologic treatment in solid malignancies–not an innocent bystander. Critical Reviews in Oncology/hematology. 2018 Jun 1;126:37-44.
29. Flaherty RL, Intabli H, Falcinelli M, Bucca G, Hesketh A, Patel BA, Allen MC, Smith CP, Flint MS. Stress hormone-mediated acceleration of breast cancer metastasis is halted by inhibition of nitric oxide synthase. Cancer Letters. 2019 Sep 10;459:59-71.
30. Flaherty RL, Intabli H, Falcinelli M, Bucca G, Hesketh A, Patel BA, Allen MC, Smith CP, Flint MS. Stress hormone-mediated acceleration of breast cancer metastasis is halted by inhibition of nitric oxide synthase. Cancer Letters. 2019 Sep 10;459:59-71.
31. Kassi E, Moutsatsou P. Glucocorticoid receptor signaling and prostate cancer. Cancer letters. 2011 Mar 1;302(1):1-10.
32. Lu D, Sinnott JA, Valdimarsdóttir U, Fang F, Gerke T, Tyekucheva S, Fiorentino M, Lambe M, Sesso HD, Sweeney CJ, Wilson KM. Stress-related signaling pathways in lethal and nonlethal prostate cancer. Clinical Cancer Research. 2016 Feb 1;22(3):765-72.
33. Ishiguro H, Kawahara T, Zheng Y, Kashiwagi E, Li Y, Miyamoto H. Differential regulation of bladder cancer growth by various glucocorticoids: corticosterone and prednisone inhibit cell invasion without promoting cell proliferation or reducing cisplatin cytotoxicity. Cancer Chemotherapy and Pharmacology. 2014 Aug 1;74(2):249-55.
34. PZheng Y, Izumi K, Li Y, Ishiguro H, Miyamoto H. Contrary regulation of bladder cancer cell proliferation and invasion by dexamethasone-mediated glucocorticoid receptor signals. Molecular cancer therapeutics. 2012 Dec 1;11(12):2621-32.
35. Pitter KL, Tamagno I, Alikhanyan K, Hosni- Ahmed A, Pattwell SS, Donnola S, Dai C, Ozawa T, Chang M, Chan TA, Beal K. Corticosteroids compromise survival in glioblastoma. Brain. 2016 May 1;139(5):1458-71.
36. Llaguno-Munive M, Medina LA, Jurado R, Romero-Piña M, Garcia-Lopez P. Mifepristone improves chemo-radiation response in glioblastoma xenografts. Cancer Cell International. 2013 Dec;13(1):29.
37. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab plus radiotherapy–temozolomide for newly diagnosed glioblastoma. New England Journal of Medicine. 2014 Feb 20;370(8):709-22.
38. Chinot OL, Nishikawa R, Mason W, Henriksson R, Saran F, Cloughesy T, et al. Upfront bevacizumab may extend survival for glioblastoma patients who do not receive second-line therapy: an exploratory analysis of AVAglio. Neuro-oncology. 2016 Sep 1;18(9):1313-8.
39. Sau S, Banerjee R. Cationic lipid-conjugated dexamethasone as a selective antitumor agent. European Journal of Medicinal Chemistry. 2014 Aug 18;83:433-47.
40. Licarete E, Rauca VF, Luput L, Drotar D, Stejerean I, Patras L, et al. Overcoming Intrinsic Doxorubicin Resistance in Melanoma by Anti-Angiogenic and Anti- Metastatic Effects of Liposomal Prednisolone Phosphate on Tumor Microenvironment. International Journal of Molecular Sciences. 2020 Jan;21(8):2968.
41. Rathore B, Jaggarapu MM, Ganguly A, Rachamalla HK, Banerjee R. Cationic lipid-conjugated hydrocortisone as selective antitumor agent. European Journal of Medicinal Chemistry. 2016 Jan 27;108:309- 21.
42. Kim JH, Hwang YJ, Han SH, Lee YE, Kim S, Kim YJ, et al. Dexamethasone inhibits hypoxia-induced epithelial-mesenchymal transition in colon cancer. World Journal of Gastroenterology: WJG. 2015 Sep 14;21(34):9887.
43. Patras L, Sylvester B, Luput L, Sesarman A, Licarete E, Porfire A, et al. Liposomal prednisolone phosphate potentiates the antitumor activity of liposomal 5-fluorouracil in C26 murine colon carcinoma in vivo. Cancer Biology & Therapy. 2017 Aug 3;18(8):616-26.
44. Liu X, Cui H, Niu H, Wang L, Li X, Sun J, et al. Hydrocortisone Suppresses Early Paraneoplastic Inflammation And Angiogenesis To Attenuate Early Hepatocellular Carcinoma Progression In Rats. OncoTargets and Therapy. 2019;12:9481.
45. Shang F, Liu M, Li B, Zhang X, Sheng Y, Liu S, Han J, et al. The anti-angiogenic effect of dexamethasone in a murine hepatocellular carcinoma model by augmentation of gluconeogenesis pathway in malignant cells. Cancer Chemotherapy and Pharmacology. 2016 May 1;77(5):1087-96.
46. Geng Y, Wang J, Jing H, Wang HW, Bao YX. Inhibitory effect of dexamethasone on Lewis mice lung cancer cells. Genetics and Molecular Research. 2014 Jan 1;13(3):6827-36.
47. Sun N, Ji H, Wang W, Zhu Q, Cao M, Zang Q. Inhibitory effect of dexamethasone on residual Lewis lung cancer cells in mice following palliative surgery. Oncology Letters. 2017 Jan 1;13(1):356-62.
48. Busada JT, Ramamoorthy S, Cain DW, Xu X, Cook DN, Cidlowski JA. Endogenous glucocorticoids prevent gastric metaplasia by suppressing spontaneous inflammation. The Journal of Clinical Investigation. 2019 Mar 1;129(3):1345-58.
49. Min KJ, Jang JH, Lee JT, Choi KS, Kwon TK. Glucocorticoid receptor antagonist sensitizes TRAILinduced apoptosis in renal carcinoma cells through upregulation of DR5 and down-regulation of c-FLIP (L) and Bcl-2. Journal of Molecular Medicine. 2012 Mar 1;90(3):309-19.
50. Zhang C, Marmé A, Wenger T, Gutwein P, Edler L, Rittgen W, et al. Glucocorticoid-mediated inhibition of chemotherapy in ovarian carcinomas. International Journal of Oncology. 2006 Feb 1;28(2):551-8.
51. Chen YX, Wang Y, Fu CC, Diao F, Song LN, Li ZB, et al. Dexamethasone enhances cell resistance to chemotherapy by increasing adhesion to extracellular matrix in human ovarian cancer cells. Endocrine-Related Cancer. 2010 Mar 1;17(1):39.