Commentary
Chronic liver diseases with different etiologies can provoke a fibrotic wound-healing response, which leads to liver fibrosis. Liver fibrosis is characterized by abnormal deposition and distribution of extracellular matrix (ECM), which restricts the regeneration of normal liver, and finally results in liver cirrhosis, liver failure or even hepatocellular carcinoma (HCC) [1]. Etiologically, about 40% of HCC is caused by hepatitis B virus (HBV), 40% caused by hepatitis C virus (HCV), 11% caused by chronic alcohol abuse, and about 10% due to other causes, with an increasing prevalence of nonalcoholic fatty liver disease, and all these etiologies could lead to liver fibrosis, and contribute to a favorable niche for tumorgenesis [2]. Globally, liver cirrhosis currently accounts for approximately 1.16 million death each year, which ranks the 11th most common causes of death [2]. Despite increasing development of therapeutic strategies in the past two decades, there is still no approved anti-fibrotic drug to date [3]. Therefore, it is urgent to make further elucidation of the mechanism of liver fibrogenesis.
Hepatic stellate cell (HSC) activation is the central event of liver fibrosis. During this process, the quiescent vitamin- A-storing HSCs transdifferentiate into myofibroblasts, with enhanced abilities of ECM production, proliferation, contraction and migration [4]. HSC activation is regulated by various pathways and mediators, and induction of apoptosis, deactivation and senescence are the critical pathways for HSCs clearance and liver fibrosis resolution [1,5,6]. As recently reported, fibrosis is orchestrated by the ‘big five’, including macrophages, myofibroblasts, matrix, mechanics and miscommunication [7]. HSCs are resided in the ECM, and ECM not only provides structural support for HSCs, but also modulates the biological processes of HSCs [7]. Thus, investigating the role of ECM components in HSC activation and liver fibrosis might contribute to novel anti-fibrotic strategies.
Matricellular proteins are important components of ECM, and these non-structural proteins are located in the cytoplasm or secreted to the ECM, which modulate the interaction between matrix and cells, and exert crucial roles in embryo development, inflammatory response, tissue remodeling and tumor progression [8,9]. In the recent years, the role of matricellular protein in liver fibrosis has drawn great attention. For example, osteopontin expression is increased in multiple chronic liver fibrotic models, and it promotes HSC activation and collagen deposition [10-12]. As a kind of matricellular protein, Sparc/osteonectin, cwcv and kazal-like domain proteoglycan 1 (SPOCK1) belongs to the secreted protein, acidic, and rich in cysteine (SPARC) family [13]. SPOCK1 is initially identified in the human testis, thus, it is also named as Testican-1, and subsequent studies reveal that SPOCK1 is expressed in multiple tissues. The function of SPOCK1 is not fully elucidated, and it might be correlated with protease inhibition [14,15]. SPOCK1 mainly exerts oncogenic roles in various tumors by promoting proliferation, invasion and survival, including HCC, gallbladder cancer, and gastric cancer, etc [16-18]. It is widely acknowledged that liver fibrosis is a precancerous stage of HCC [19], however, the role of SPOCK1 in liver fibrogenesis remains unknown.
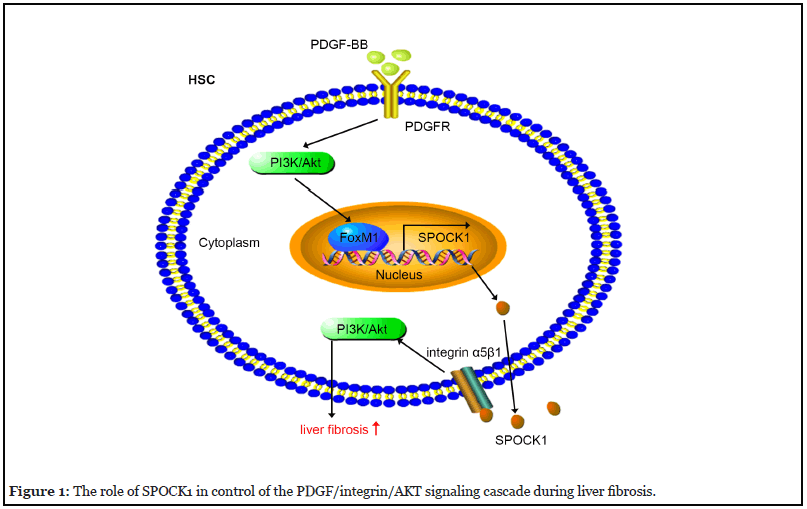
In the recent study, for the first time, we elucidated the roles, mechanisms, and implications of SPOCK1 in liver fibrosis [20]. We found that SPOCK1 expression was dramatically upregulated in human and rat fibrotic liver tissues and in activated primary rat HSCs. Subsequently, we found that SPOCK1 expression in HSCs was induced by transforming growth factor (TGF)-β1 and platelet-derived growth factor (PDGF)-BB. Interestingly, previous studies reported that SPOCK1 is a target gene of TGF-β1 in lung cancer cells and breast cancer cells [21,22]. As PDGFBB exerted the stronger effect on SPOCK1 expression, we mainly focused on the mechanism how PDGF-BB upregulated SPOCK1 expression. Our results showed that PDGF-BB promoted SPOCK1 expression through activating the phosphatidylinositol 3-kinase/protein kinase B/ forkhead box M1 (PI3K/Akt/FoxM1) signaling pathway. It is widely acknowledged that PDGF-BB promotes HSC activation, proliferation and migration [23]. We found that, as a target gene of PDGF-BB, SPOCK1 was involved in PDGF-BB-induced pro-fibrotic responses, and SPOCK1 promoted HSCs activation, proliferation and migration by activating the PI3K/Akt signaling pathway. Interestingly, there might exist a positive feedback loop between PI3K/ Akt activation and SPOCK1 expression, that is, PDGF-BB upregulates SPOCK1 expression through activating the PI3K/Akt pathway, and SPOCK1 reciprocally activates the PI3K/Akt pathway, thus resulting in an augmentation of the pro-fibrotic responses.
Mechanistically, most matricellular proteins exert their roles by interacting with integrins, and subsequently modulate the intracellular signaling cascades, and therefore regulate the transformation of biological processes. Integrins are a class of heterodimeric cell surface receptors, to date, integrin α1β1, α2β1, α5β1, α6β4, α8β1, αvβ1, αvβ3, αvβ5, αvβ6, αvβ8, α8β1, and α11β1 are found to be expressed in HSCs and exert important roles in liver fibrogenensis [24,25]. For instance, osteopontin activates the PI3K/Akt/nuclear factor kappa-B (NF-κB) signaling pathway and upregulates collagen-1 expression by interacting with integrin αvβ3 [12]; while periostin promotes collagen deposition and HSC migration by interacting with integrin αvβ3 and integrin αvβ5 [26]. These evidences drive us to propose whether SPOCK1 exerts its pro-fibrotic roles through interaction with integrins. By coimmunoprecipitation and immunofluorescence, we found that SPOCK1 interacted with integrin α5β1. Furthermore, blockade of integrin α5β1 by neutralizing antibodies abrogated recombinant SPOCK1-induced activation, proliferation, migration of HSCs and activation of PI3K/ Akt signaling pathway.
Finally, we performed in vivo assays to verify the role of SPOCK1 expressed by HSCs in liver fibrogenesis. As previously reported, liver fibrogenesis is orchestrated by several cells including hepatocytes, HSCs, macrophages, etc., and different cells may exert different or even opposite effects [27]. For instance, apoptosis of hepatocytes is the predominant initiator of liver fibrogenesis, while apoptosis of HSCs is a critical pathway for resolution of liver fibrosis [1]. As HSCs is the central effector for collagen production, it is necessary and important to achieve HSC-specific gene delivery when performing in vivo assays to explore the role of a certain gene in liver fibrogenesis. In general, most in vivo studies utilized regular lentivirus, adenovirus or recombinant adeno-associated virus (rAAV), however, these viruses have no cell specificity, and after injected via tail vein, they infected all the cells in liver [28-30]. Under these circumstances, it is difficult to clarify which cell exerts the predominant role, furthermore, these strategies might lead to undesired side effects. In the recent years, two methods of HSC-specific gene delivery have come into spotlight. Firstly, vitamin A-coupled liposomes are used to deliver small interfering RNA (siRNA) to achieve HSCspecific gene knockdown [30]. Secondly, a lentiviral or rAAV construct is established, in which the targeted sequence is cloned downstream of the promoter of glial fibrillary acidic protein (GFAP) or α-smooth muscle actin (α-SMA) (two HSCs markers), and then, the construct is injected through tail vein to achieve HSC-specific gene delivery [30,31]. In our study, we utilized the second method, a HSC-targeted lentivirus (LV-SMA-shSPOCK1-Flag) was constructed and injected via tail vein, and the rat liver fibrotic model was established by intraperitoneal injection of thioacetamide (TAA). The results showed that HSC-specific knockdown of SPOCK1 significantly alleviated TAA-induced rat liver fibrosis. Taken together, SPOCK1 is upregulated in liver fibrosis, and its overexpression is mediated by PDGF-BB through activating PI3K/Akt/FoxM1 pathway; SPOCK1 is involved in PDGF-BB-induced pro-fibrotic responses by interacting with integrin α5β1 and activating the PI3K/ Akt pathway; finally, HSC-specific knockdown of SPOCK1 ameliorates liver fibrosis in vivo.
It is widely acknowledged that TGF-β1 and PDGF-BB are two most important pro-fibrotic cytokines [27]. However, TGF-β1 and PDGF-BB are ubiquitously expressed in all organs, and play critical roles in cell proliferation, differentiation, and maintenance of physiological function. Unfortunately, direct blockade of TGF-β1 or PDGFBB could result in severe side effects; therefore, these strategies are unfavorable for liver fibrosis treatment [32-34]. Interestingly, our results showed that SPOCK1 might be the common downstream target of TGF-β1 and PDGFBB, although further studies are needed to elucidate how TGF-β1 promoted SPOCK1 expression. From our perspective, the most appealing contribution of our study is that targeting SPOCK1 may block part of the pathways of both TGF-β1 and PDGF-BB at the same time, and thereby alleviating liver fibrosis with fewer side effects. Therefore, it is noteworthy that SPOCK1 might be a potential pharmacological target for liver fibrosis treatment, which needs further clinical study. Furthermore, it remains to determine whether neutralizing antibodies of integrin α5β1 could ameliorate liver fibrosis in rats/mice, or even applicable for clinical treatment of liver fibrosis.
In summary, our study uncovered the role, mechanism, and implication of SPOCK1 in liver fibrosis, indicating that targeting SPOCK1/integrin α5β1 axis might be an effective and safe therapeutic approach for liver fibrosis.
Conflict of Interest
The author declared no conflict of interest.
Funding
This study was financially supported by National Natural Science Foundation of China (No.81772610, 81974071).
References
2. Asrani, SK, Devarbhavi, H, Eaton, J, Kamath, PS. Burden of liver diseases in the world. J Hepatol.2019;70(1):151-171.
3. Lemoinne, S, Friedman, SL. New and emerging anti-fibrotic therapeutics entering or already in clinical trials in chronic liver diseases. Curr Opin Pharmacol. 2019;(49):60-70.
4. Fan Y, Du Z, Steib CJ, Ding Q, Lu P, Tian D, et al. Effect of SEPT6 on the biological behavior of hepatic stellate cells and liver fibrosis in rats and its mechanism. Lab Invest. 2019;99(1):17-36.
5. Troeger JS, Mederacke I, Gwak GY, Dapito DH, Mu X, Hsu CC, et al. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology. 2012;143(4):1073-1083 e1022.
6. Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A. 2012 Jun 12;109(24):9448-53.
7. Pakshir P, Hinz B. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. 2018;(68-69):81-93.
8. Murphy-Ullrich, JE, Sage, EH. Revisiting the matricellular concept. Matrix Biol. 2014;(37):1-14.
9. Wu T, Ouyang G. Matricellular proteins: multifaceted extracellular regulators in tumor dormancy. Protein Cell. 2014;5(4):249-252.
10. Wang X, Lopategi A, Ge X, Lu Y, Kitamura N, Urtasun R, et al. Osteopontin induces ductular reaction contributing to liver fibrosis. Gut. 2014;63(11):1805-1818.
11. Lorena D, Darby IA, Gadeau AP, Leen LL, Rittling S, Porto LC, et al. Osteopontin expression in normal and fibrotic liver. altered liver healing in osteopontin-deficient mice. J Hepatol. 2006;44(2):383-390.
12. Urtasun R, Lopategi A, George J, Leung TM, Lu Y, Wang X, et al. Osteopontin, an oxidant stress sensitive cytokine, up-regulates collagen-I via integrin alpha(V) beta(3) engagement and PI3K/pAkt/NFkappaB signaling. Hepatology. 2012;55(2):594-608.
13. Bradshaw AD. Diverse biological functions of the SPARC family of proteins. Int J Biochem Cell Biol.2012;44(3):480-488.
14. Bocock JP, Edgell CJ, Marr HS, Erickson AH. Human proteoglycan testican-1 inhibits the lysosomal cysteine protease cathepsin L. Eur J Biochem. 2003;270(19):4008-4015.
15. Edgell CJ, BaSalamah MA, Marr HS. Testican-1: a differentially expressed proteoglycan with protease inhibiting activities. Int Rev Cytol. 2004;236:101-122.
16. Li Y, Chen L, Chan TH, Liu M, Kong KL, Qiu JL, et al. SPOCK1 is regulated by CHD1L and blocks apoptosis and promotes HCC cell invasiveness and metastasis in mice. Gastroenterology. 2013;144(1):179-191 e174.
17. Shu YJ, Weng H, Ye YY, Hu YP, Bao RF, Cao Y, et al. SPOCK1 as a potential cancer prognostic marker promotes the proliferation and metastasis of gallbladder cancer cells by activating the PI3K/AKT pathway. Mol Cancer. 2015;14(1):12.
18. Chen D, Zhou H, Liu G, Zhao Y, Cao G, Liu Q. SPOCK1 promotes the invasion and metastasis of gastric cancer through Slug-induced epithelial-mesenchymal transition. J Cell Mol Med. 2018;22(2):797-807.
19. El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118-1127.
20. Du Z, Lin Z, Wang Z, Liu D, Tian D, Xia L. SPOCK1 overexpression induced by platelet-derived growth factor-BB promotes hepatic stellate cell activation and liver fibrosis through the integrin alpha5beta1/PI3K/Akt signaling pathway. Lab Invest. 2020;100(8):1042-1056.
21. Miao L, Wang Y, Xia H, Yao C, Cai H, Song Y. SPOCK1 is a novel transforming growth factor-beta target gene that regulates lung cancer cell epithelialmesenchymal transition. Biochem Biophys Res Commun. 2013;440(4):792-797.
22. Fan LC, Jeng YM, Lu YT, Lien HC. SPOCK1 Is a Novel Transforming Growth Factor-beta-Induced Myoepithelial Marker That Enhances Invasion and Correlates with Poor Prognosis in Breast Cancer. PLoS One. 2016;11(9):e0162933.
23. Shah R, Reyes-Gordillo K, Rojkind M. Thymosin beta4 inhibits PDGF-BB induced activation, proliferation, and migration of human hepatic stellate cells via its actinbinding domain. Expert Opin Biol Ther. 2018;18(sup1):177-184.
24. Schuppan D, Ashfaq-Khan M, Yang AT, Kim YO. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018;(68-69):435-451.
25. Schnittert J, Bansal R, Storm G, Prakash J. Integrins in wound healing, fibrosis and tumor stroma: High potential targets for therapeutics and drug delivery. Adv Drug Deliv Rev. 2018;(129):37-53.
26. Sugiyama A, Kanno K, Nishimichi N, Ohta S, Ono J, Conway SJ, et al. Periostin promotes hepatic fibrosis in mice by modulating hepatic stellate cell activation via alphav integrin interaction. J Gastroenterol. 2016;51(12):1161- 1174.
27. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14(7):397-411.
28. Zhang K, Han Y, Hu Z, Zhang Z, Shao S, Yao Q, et al. SCARNA10, a nuclear-retained long non-coding RNA, promotes liver fibrosis and serves as a potential biomarker. Theranostics. 2019;9(12):3622-3638.
29. Pan XY, You HM, Wang L, Bi YH, Yang Y, Meng HW, et al. Methylation of RCAN1.4 mediated by DNMT1 and DNMT3b enhances hepatic stellate cell activation and liver fibrogenesis through Calcineurin/NFAT3 signaling. Theranostics. 2019;9(15):4308-4323.
30. Wu X, Wu X, Ma Y, Shao F, Tan Y, Tan T, et al. CUG-binding protein 1 regulates HSC activation and liver fibrogenesis. Nat Commun. 2016;(7):13498.
31. Kim KM, Han CY, Kim JY, Cho SS, Kim YS, Koo JH, et al. Galpha12 overexpression induced by miR-16 dysregulation contributes to liver fibrosis by promoting autophagy in hepatic stellate cells. J Hepatol. 2018;68(3):493-504.
32. Dewidar B, Meyer C, Dooley S, Meindl-Beinker AN. TGF-beta in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells. 2019;8(11):1419.
33. Srikanthan A, Ethier JL, Ocana A, Seruga B, Krzyzanowska MK, Amir E. Cardiovascular toxicity of multi-tyrosine kinase inhibitors in advanced solid tumors: a population-based observational study. PLoS One. 2015;10(3):e0122735.
34. Chrisoulidou A, Mandanas S, Margaritidou E, Mathiopoulou L, Boudina M, Georgopoulos K, et al. Treatment compliance and severe adverse events limit the use of tyrosine kinase inhibitors in refractory thyroid cancer. Onco Targets Ther. 2015;(8):2435-2442.