Abstract
Sepsis is a systemic inflammatory response caused by a harmful host immune reaction that is activated in response to microbial infections. Infection-induced type I interferons (IFNs) play critical roles during septic shock. Type I IFNs initiate their biological effects by binding to their transmembrane interferon receptors and initiating the phosphorylation and activation of tyrosine kinases TYK2 and JAK1, which promote phosphorylation and activation of STAT molecules. Type I IFN-induced activation of JAK/STAT pathways is a complex process and not well understood. Improper regulation of type I IFN responses can lead to the development of infectious and inflammation-related diseases, including septic shock, autoimmune diseases, and inflammatory syndromes. This review is mainly focused on the possible mechanistic roles of the transmembrane Perforin-2 molecule in the regulation of type I IFNinduced signaling.
Keywords
JAK/STAT, Interferons, TYK2, STATs, Perforin-2, IFNAR1, IFNAR2 and Signaling
Introduction
Type I interferons (IFNs) produced in response to infections with either bacterial, viral or fungal pathogens are critical to induce antimicrobial defenses in infected and adjacent cells to reduce the spread of microbes, initiate communication between innate and adaptive immunity, and to promote the survival of the host [1]. Improper induction or regulation of type I IFN signaling can lead to chronic infection-induced inflammation-related diseases, including septic shock, autoimmune diseases, and inflammatory syndromes [2].
Type I IFNs consist of a group of structurally similar cytokines including IFN-α (which can be further subdivided into 13 different subtypes), IFN-β, IFN-δ, IFN-ε, IFN-κ, IFN-τ, and IFN-ω1–3. These IFNs can act in an autocrine, paracrine, or systemic fashion. Studies have shown that all type I IFNs exclusively bind to and signal through ubiquitously expressed heterodimeric transmembrane (TM) receptors composed of two subunits—IFNAR1 and IFNAR2 [3], which are constitutively associated with tyrosine kinases TYK2 and JAK1/STAT2 (signal transducer and activator of transcription 2), respectively, under normal physiological conditions [4,5]. Upon binding of type I IFNs to IFNAR1 and IFNAR2, the proximal receptor complex is assembled and the tyrosine kinases TYK2 and JAK1 are activated by reciprocal transphosphorylation. Although activated TYK2 and JAK1 phosphorylate and activate STAT2, which is a critical early step for STAT1 activation by type I IFN in canonical signaling [6], the mechanism of STAT2-dependent STAT1 activation after stimulation of type I IFN is poorly understood. Activated STAT1 and STAT2 monomers dimerize and interact with IRF9 (IFN-regulatory factor 9) in the cytoplasm, which is known as the ISGF3 (IFN-stimulated gene (ISG) factor 3) complex [1,7]. This ISGF3 complex translocates to the nucleus, binds IFN-stimulated response elements (ISREs) in DNA, and initiates the activation of several hundred interferon stimulated genes (ISGs) [8,9]. Type I IFN– activated STAT1 can also form a homodimer known as IFNγ-Activated Factor (GAF), which migrates into the nucleus, and binds to IFNγ-Activated Sequences (GAS), to induce proinflammatory genes [10,11]. Type I IFN activated STAT1 and STAT2 also form other heterodimers, including STAT1-STAT3, STAT1-STAT4, STAT1-STAT5, and STAT2-STAT3 in specific cell types such as endothelial cells or cells of lymphoid origin [3,12-14].
Non-canonical or alternative type I IFN signaling can activate additional pathways such as Crk-like protein (CrkL), the phosphatidyl-inositol 3-kinase (PI3K) pathway, and the mitogen-activated protein kinase (MAPK) pathway [15]. The type I IFN-induced CrkL pathway is critical for the proliferation of both hematopoietic and non-hematopoietic cells. STAT5, which is constitutively associated with IFNAR1 and phosphorylated by TYK2 upon stimulation with type I IFN, forms a complex with CrkL [15,16]. This STAT5-CrkL complex translocates into the nucleus, binds to GAS elements, and induces growthinhibitory genes [17,18]. The Type I IFN-induced PI3K pathway is involved in the production of antiviral effectors and in the regulation of the transcription and translation of ISGs [19]. Type I IFN-activated JAK1/TYK2 also induces rapid phosphorylation and activation of insulin receptor substrate 1 (IRS1) and 2 (IRS2), which subsequently bind to the catalytic p85 subunit of PI3K, which is required for the activation of the regulatory p110 subunit of PI3K [20]. Type I IFN-induced activation of the p85 subunit of PI3K is critical for Akt-dependent mammalian target of rapamycin (mTOR) activation and mRNA translation [21]. Type I IFN-induced JAKs also mediate the activation of the small GTPase, Rac1, and guanine-nucleotide-exchange (GEF) factor VAV [22-24], which are required for the activation of extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK) and p38 MAP kinases [25-27]. Type I IFN-induced activation of p38/ERK and MAPK pathways play antiviral roles [25,28-30].
Perforin-2, also known as macrophage-expressed gene 1 (MPEG1), is a major anti-bacterial effector protein of the innate immune system in both phagocytic and tissue forming cells [31-33]. The expression of Perforin-2 is not limited to macrophages but is found in many other cell types, including murine embryonic fibroblasts (MEFs) and epithelial cells [34]. It is a ~72-kDa protein localized in select intracellular membranes and at the cell surface [34,35]. Perforin-2 forms pores in grampositive, gram-negative, and acid-fast bacteria and limits the proliferation and spread of these bacteria [36,37]. Perforin-2 is highly conserved throughout evolution from sea sponges to humans [31]. Some studies have also suggested that Perforin-2 may have an anti-microbial role in invertebrates, including clams, mussels, and snails [35]. Evolutionary studies of Perforin-2 have demonstrated that it is one of the oldest eukaryotic MACPF members, present in early metazoan phyla, including Porifera (sponges) [31]. Perforin-2 contains an N-terminal Membrane Attack Complex/Perforin (MACPF) domain, a Perforin-2 (P2) domain, a single transmembrane domain (TM), and a C-terminal cytoplasmic tail (CT) containing a FERMBinding motif [31,34]. There is also a shorter, alternatively spliced Perforin-2 form that lacks the TM and CT domains, but its function is not well understood [38].
Regulation of IFNAR1 and IFNAR1 activation by Perforin-2
Perforin-2 has recently been shown to play additional roles in innate immunity to bacterial infections [34]. The stimulation of Toll-like receptor-4 (TLR-4) by the central outer membrane molecule of gram-negative bacteria, lipopolysaccharide (LPS), initiates the production of type I interferons through the TRIF adaptor molecule [39,40]. LPS-induced type I interferons play critical roles in LPS-induced toxic shock [41]. Perforin-2 knockout mice challenged with a high dose of LPS were resistant to LPS-induced septic shock [34]. This phenomenon was similar to mice deficient in IFNAR1 or TYK-2, which are critical effectors of type I IFN signaling [42-44]. Perforin-2 knockout macrophages also demonstrated decreased production of IP-10 (CXCL10) after LPS challenge [34]. IP-10 is induced by type I IFN in a STAT1-dependent manner and plays a critical role in LPS-induced sepsis [45-47]. Perforin-2 is also vital for type I-IFN–mediated inhibition of viral replication [34]. These findings prompted the authors to investigate whether Perforin-2 plays a role in type I IFN signaling. Interestingly, the authors found that Perforin-2 was expressed on the cell surface as an integral component of type I IFN signaling. Both IFNAR1 and IFNAR2 are expressed on the cell surface in an isolated form in resting cells [5,48]. IFNAR1 and IFNAR2 rapidly dimerize upon stimulation with type I IFN to activate downstream signaling events; however, the mechanism that brings these receptors together after type I IFN stimulation is poorly understood. Intriguingly, the N-linked glycosylated extracellular MACPF domain of Perforin-2 is constitutively associated with IFNAR1, known as ligand-independent complex I, in resting cells (Figure 1). Similarly, the N-linked glycosylated extracellular P2 domain of Perforin-2 is constitutively associated with IFNAR2, known as ligand-independent complex II, in resting cells (Figure 1). In resting cells, Perforin-2 may be keeping IFNAR1 and IFNAR2 in the correct conformation, which is crucial to forming an active ligand dependent complex III essential for downstream signaling [34]. Loss of N-linked glycosylation in either the MACPF or P2 domains of Perforin-2 resulted in loss of complex I and II, respectively, as well as active complex III after stimulation with type I IFN and downstream signaling (Figure 1) [34]. Although the extracellular MACPF and P2 domains of Perforin-2 are constitutively associated with IFNAR1 and IFNAR2, the extracellular domain/motif of IFNAR1 and IFNAR2 that interact with MACPF and P2 domains are not known [34].
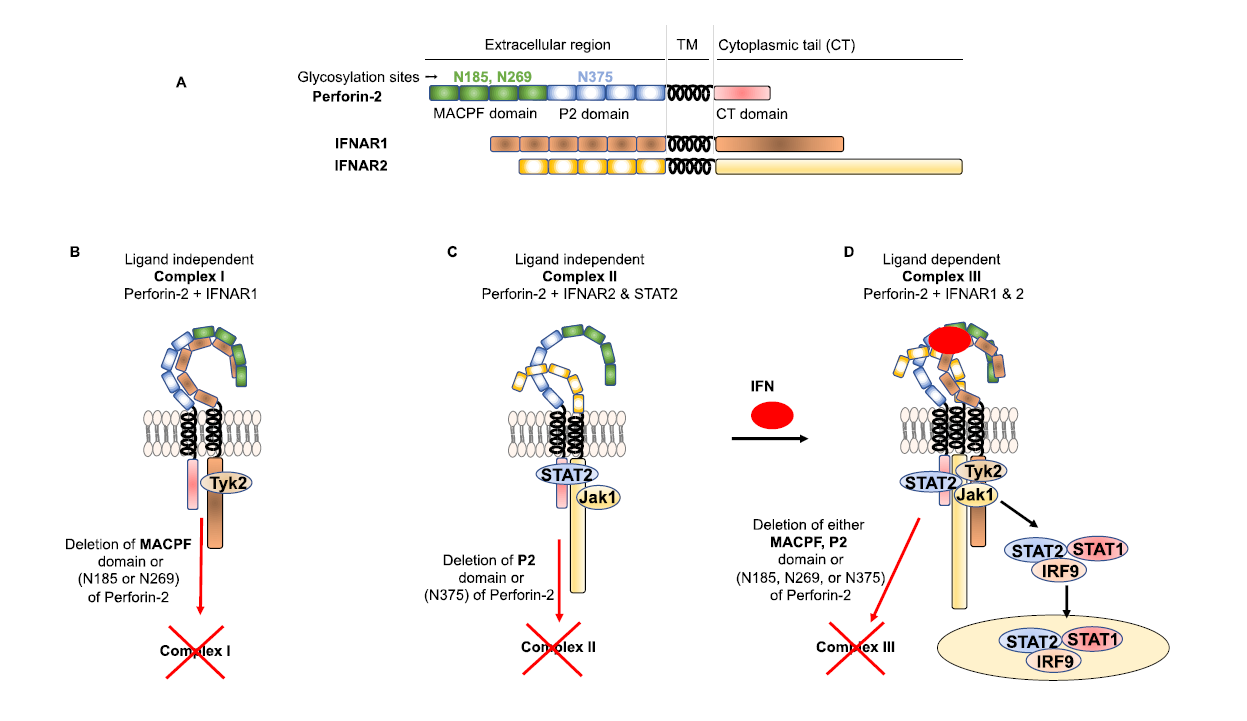
Regulation of TYK2 and JAK1 phosphorylation and activation by Perforin-2
The multidomain non-receptor tyrosine kinases TYK2 and JAK1 belong to the JAK family in mammals [49]. Many cytokines and growth factor receptors regulate the activity of intracellular JAKs and vice versa in the immune system to limit harmful stimuli, microbial infections, and inflammatory diseases. Deletion or loss of TYK2 or JAK1 leads to severely compromised immune function [50,51]. JAK1 and TYK2 constitutively and non-covalently associate with their cognate signaling receptors in resting cells [52]. JAK1 and TYK2 share conserved N-terminal FERM and SH2 domains, followed by a pseudokinase and a kinase domain. FERM and SH2 domains are essential for association with the cytoplasmic domain of cytokine receptors [53]. The kinase domain is critical for activating downstream signaling, while the pseudokinase domain suppresses the kinase activity of JAK1 and TYK2 [54-57]. Upon stimulation with type I IFN, the heterodimeric receptor complex, IFNAR1 and IFNAR2, is formed and phosphorylated, activating JAK1 and TYK2 phosphorylation required for downstream signaling [3].
Although the reciprocal transphosphorylation of JAK1 and TYK2 is impaired in the absence of Perforin-2 expression after stimulation with type I IFN, the constitutive interactions between IFNAR2-JAK1 and TYK2-IFNAR1 are not affected [34]. While the interactions of the MACPF and P2 domains of Perforin-2 with IFNAR1 and IFNAR2, respectively, are essential for phosphorylation and activation of JAK1 and TYK2 after stimulation with type I IFN, the phosphorylation and activation of JAK1 and TYK2 were also impaired in the absence of the CT domain of Perforin-2 [34]. These findings suggest that the CT domain of Perforin-2 regulates the reciprocal transphosphorylation of JAK1 and TYK2 after stimulation with type I IFN [34]. However, the mechanism by which the CT domain mediates reciprocal transphosphorylation of JAK1 and TYK2 after stimulation with type I IFN is unknown.
Regulation of STAT1 and STAT2 phosphorylation and activation by perforin-2
Activation of the STAT family of transcription factors in response to stimulation with type I IFN is critical for the induction of specific genes involved in antiviral, antitumor, and anti-proliferative activities [58]. Dysregulation of STAT activation after stimulation with type I IFN could lead to autoimmune disorders such as Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis, and systemic sclerosis [58,59]. There are seven STAT family members (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b and STAT6) that bind to GAS elements [60]. In contrast, the trimeric (ISGF3) complex containing STAT1, STAT2 and IRF9 binds to ISREs [61]. All members of the STAT family are composed of seven domains (N-terminal, coiled-coil, DNA binding, linker, SH2, transactivation, and C-terminal) [9,62]. Most of the STAT proteins interact with the cytoplasmic tail of receptors [63-65]. The tyrosine phosphorylation and activation of STAT1 and STAT2 are induced after stimulation with type I IFN; however, STAT1 and STAT2 phosphorylation and activation after stimulation with type I IFN are severely inhibited in Perforin-2-deficient cells [34]. Studies have shown that STAT2 is constitutively associated with the cytoplasmic tail of IFNAR2 [4]. Surprisingly, the CT domain of Perforin-2 is critical for STAT2’s constitutive interaction with the cytoplasmic tail of IFNAR2 [34]. However, the CT domain of Perforin-2 alone does not facilitate the recruitment of STAT2 to IFNAR2 in the absence of the P2 domaindirected association of Perforin-2 with IFNAR2 [34].
Regulation of non-canonical type I IFN signaling by Perforin-2
The canonical type I IFN-induced ISGF33 complex is controlled by activated STAT3 [66]. Activated STAT3 forms a complex with STAT1 (STAT1:STAT3), but not STAT2 or IRF9, and destabilizes the ISGF3 complex [67,68]. However, whether Perforin-2 is required for the activation of STAT3 is not known. Studies have shown that STAT4 is constitutively associated with IFNAR2 and is activated by type I IFN [69]. Similarly, STAT5 constitutively interacts with TYK-2 and IFNAR1 [17]; however; it is not known whether the CT domain of Perforin-2 also regulates the interaction between STAT4 and IFNAR2 or STAT5, TYK-2 and IFNAR1, respectively. TYK2-mediated STAT5 phosphorylation creates a docking site for CrkL upon stimulation with type I IFN [70]. Once associated, the STAT5:CrkL complex translocates into the nucleolus and binds to GAS elements to induce growth-inhibitory genes [15,17,71]. Similarly, activation of STAT6 after type I IFN stimulation allows STAT6 to form a complex with STAT2, which induces anti-proliferative genes [72,73]. However, it is unknown if Perforin-2 is involved in the activation of STAT5 and STAT6. The activation of PI3K and MAPK after type I IFN stimulation is significantly delayed in Perforin-2 deficient cells [34]; however; it is not clear whether Perforin-2 is directly regulating the activation of PI3K subunits, p85 or p110, or PI3K activation of IRS1 or IRS2.
Conclusion
In the past several years, most of the work on type I IFN signaling has been focused on determining the molecular mechanisms of type I IFN-mediated JAK-STAT signaling. Recent work has demonstrated how type I IFN-mediated canonical and non-canonical signaling could be regulated by Perforin-2. While more work is needed to characterize the precise mechanisms by which Perforin-2 may be regulating type I IFN signaling at different checkpoints, we have already confirmed a primary role for Perforin-2 in type I IFN-mediated signaling.
Based on recent findings, it is clear that Perforin-2 is playing an important role in regulating the type I IFN response and protecting the host from viral infection, but a number of issues remain to be addressed. Although a low level of Perforin-2 expression is sufficient for the activation of type I IFN signaling, what is the role of constitutively high expression of Perforin-2 in immune cells such as macrophages and neutrophils in the context of type I IFN signaling? In addition, due to the constitutive association of Perforin-2 with IFNAR1 and IFNAR2, it will be important to develop highly selective inhibitors targeting the Perforin-2-IFNAR1 and Perforin-2-IFNAR2 complexes in order to elucidate the roles of these complexes in tissue homeostasis.
Authors contribution
G. V. P., and N. S. wrote and edited the review.
Funding
This work was supported by American Cancer Society Grant RSG-16-254-01-MPC (to N.S.).
Conflicts of Interest
The authors declare no conflict of interest.
References
2. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015;15:87-103.
3. Platanias LC. Mechanisms of type-I- and type-IIinterferon- mediated signalling. Nat Rev Immunol.2005;5:375-86.
4. Li X, Leung S, Kerr IM, Stark GR. Functional subdomains of STAT2 required for preassociation with the alpha interferon receptor and for signaling. Mol Cell Biol. 1997;17:2048-56.
5. Schreiber G, Piehler J. The molecular basis for functional plasticity in type I interferon signaling. Trends in Immunology. 2015;36:139-49.
6. Majoros A, Platanitis E, Kernbauer-Holzl E, Rosebrock F, Muller M, Decker T. Canonical and Non-Canonical Aspects of JAK-STAT Signaling: Lessons from Interferons for Cytokine Responses. Frontiers in Immunology. 2017;8:29.
7. Hall JC, Rosen A. Type I interferons: crucial participants in disease amplification in autoimmunity. Nature Reviews Rheumatology. 2010;6:40-9.
8. Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. 2007;6:975-90.
9. Levy DE, Darnell JE, Jr. Stats: transcriptional control and biological impact. Nature Reviews Molecular Cell Biology. 2002;3:651-62.
10. Hu X, Herrero C, Li WP, Antoniv TT, Falck-Pedersen E, Koch AE, et al. Sensitization of IFN-gamma Jak- STAT signaling during macrophage activation. Nature Immunology. 2002;3:859-66.
11. Tassiulas I, Hu X, Ho H, Kashyap Y, Paik P, Hu Y, et al. Amplification of IFN-alpha-induced STAT1 activation and inflammatory function by Syk and ITAM-containing adaptors. Nature Immunology. 2004;5:1181-9.
12. Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annual Review of Immunology. 2005;23:307-36.
13. O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. The New England Journal of Medicine. 2013;368:161-70.
14. Stark GR, Cheon H, Wang Y. Responses to Cytokines and Interferons that Depend upon JAKs and STATs. Cold Spring Harbor Perspectives in Biology. 2018;10.
15. Stanifer ML, Pervolaraki K, Boulant S. Differential Regulation of Type I and Type III Interferon Signaling. Int J Mol Sci. 2019;20.
16. Uddin S, Lekmine F, Sassano A, Rui H, Fish EN, Platanias LC. Role of Stat5 in type I interferon-signaling and transcriptional regulation. Biochemical and Biophysical Research Communications. 2003;308:325-30.
17. Fish EN, Uddin S, Korkmaz M, Majchrzak B, Druker BJ, Platanias LC. Activation of a CrkL-stat5 signaling complex by type I interferons. The Journal of Biological Chemistry. 1999;274:571-3.
18. Able AA, Burrell JA, Stephens JM. STAT5-Interacting Proteins: A Synopsis of Proteins that Regulate STAT5 Activity. Biology (Basel). 2017;6.
19. Platanias LC, Fish EN. Signaling pathways activated by interferons. Experimental Hematology. 1999;27:1583- 92.
20. Uddin S, Fish EN, Sher DA, Gardziola C, White MF, Platanias LC. Activation of the phosphatidylinositol 3-kinase serine kinase by IFN-alpha. Journal of Immunology. 1997;158:2390-7.
21. Kaur S, Sassano A, Joseph AM, Majchrzak-Kita B, Eklund EA, Verma A, et al. Dual regulatory roles of phosphatidylinositol 3-kinase in IFN signaling. Journal of Immunology. 2008;181:7316-23.
22. Platanias LC, Sweet ME. Interferon alpha induces rapid tyrosine phosphorylation of the vav proto-oncogene product in hematopoietic cells. The Journal of Biological Chemistry. 1994;269:3143-6.
23. Uddin S, Lekmine F, Sharma N, Majchrzak B, Mayer I, Young PR, et al. The Rac1/p38 mitogen-activated protein kinase pathway is required for interferon alpha-dependent transcriptional activation but not serine phosphorylation of Stat proteins. The Journal of Biological Chemistry. 2000;275:27634-40.
24. Micouin A, Wietzerbin J, Steunou V, Martyre MC. p95(vav) associates with the type I interferon (IFN) receptor and contributes to the antiproliferative effect of IFN-alpha in megakaryocytic cell lines. Oncogene. 2000;19:387-94.
25. Uddin S, Majchrzak B, Woodson J, Arunkumar P, Alsayed Y, Pine R, et al. Activation of the p38 mitogenactivated protein kinase by type I interferons. The Journal of Biological Chemistry. 1999;274:30127-31.
26. Katsoulidis E, Li Y, Mears H, Platanias LC. The p38 mitogen-activated protein kinase pathway in interferon signal transduction. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research. 2005;25:749-56.
27. Zhao LJ, Hua X, He SF, Ren H, Qi ZT. Interferon alpha regulates MAPK and STAT1 pathways in human hepatoma cells. Virol J. 2011;8:157.
28. Kaur S, Platanias LC. IFN-beta-specific signaling via a unique IFNAR1 interaction. Nature Immunology. 2013;14:884-5.
29. Mayer IA, Verma A, Grumbach IM, Uddin S, Lekmine F, Ravandi F, et al. The p38 MAPK pathway mediates the growth inhibitory effects of interferon-alpha in BCR-ABLexpressing cells. The Journal of Biological Chemistry. 2001;276:28570-7.
30. Ishida H, Ohkawa K, Hosui A, Hiramatsu N, Kanto T, Ueda K, et al. Involvement of p38 signaling pathway in interferon-alpha-mediated antiviral activity toward hepatitis C virus. Biochemical and Biophysical Research Communications. 2004;321:722-7.
31. McCormack R, Podack ER. Perforin-2/Mpeg1 and other pore-forming proteins throughout evolution. Journal of Leukocyte Biology. 2015;98:761-8.
32. Gayle P, McGaughey V, Hernandez R, Wylie M, Colletti RC, Nguyen KL, et al. Maternal- and Fetal-Encoded Perforin-2 Limits Placental Infection by a Bloodborne Pathogen. Journal of Immunology. 2020;205:1878-85.
33. McCormack R, Bahnan W, Shrestha N, Boucher J, Barreto M, Barrera CM, et al. Perforin-2 Protects Host Cells and Mice by Restricting the Vacuole to Cytosol Transitioning of a Bacterial Pathogen. Infection and Immunity. 2016;84:1083-91.
34. McCormack R, Hunte R, Podack ER, Plano GV, Shembade N. An Essential Role for Perforin-2 in Type I IFN Signaling. Journal of Immunology. 2020;204:2242- 56.
35. McCormack RM, de Armas LR, Shiratsuchi M, Fiorentino DG, Olsson ML, Lichtenheld MG, et al. Perforin-2 is essential for intracellular defense of parenchymal cells and phagocytes against pathogenic bacteria. ELife. 2015;4.
36. McCormack RM, Lyapichev K, Olsson ML, Podack ER, Munson GP. Enteric pathogens deploy cell cycle inhibiting factors to block the bactericidal activity of Perforin-2. ELife. 2015;4.
37. Ni T, Jiao F, Yu X, Aden S, Ginger L, Williams SI, et al. Structure and mechanism of bactericidal mammalian perforin-2, an ancient agent of innate immunity. Science Advances. 2020;6:eaax8286.
38. Xiong P, Shiratsuchi M, Matsushima T, Liao J, Tanaka E, Nakashima Y, et al. Regulation of expression and trafficking of perforin-2 by LPS and TNF-alpha. Cell Immunol. 2017;320:1-10.
39. Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nature Immunology. 2008;9:361-8.
40. Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145-51.
41. Thomas KE, Galligan CL, Newman RD, Fish EN, Vogel SN. Contribution of interferon-beta to the murine macrophage response to the toll-like receptor 4 agonist, lipopolysaccharide. The Journal of Biological Chemistry. 2006;281:31119-30.
42. de Weerd NA, Vivian JP, Nguyen TK, Mangan NE, Gould JA, Braniff SJ, et al. Structural basis of a unique interferon-beta signaling axis mediated via the receptor IFNAR1. Nature Immunology. 2013;14:901-7.
43. Karaghiosoff M, Steinborn R, Kovarik P, Kriegshauser G, Baccarini M, Donabauer B, et al. Central role for type I interferons and TYK2 in lipopolysaccharide-induced endotoxin shock. Nature Immunology. 2003;4:471-7.
44. Kamezaki K, Shimoda K, Numata A, Matsuda T, Nakayama K, Harada M. The role of TYK2, Stat1 and Stat4 in LPS-induced endotoxin signals. International Immunology. 2004;16:1173-9.
45. Ohmori Y, Hamilton TA. Requirement for STAT1 in LPS-induced gene expression in macrophages. Journal of Leukocyte Biology. 2001;69:598-604.
46. Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, et al. TLR4, but not TLR2, mediates IFN-betainduced STAT1alpha/beta-dependent gene expression in macrophages. Nature Immunology. 2002;3:392-8.
47. Kelly-Scumpia KM, Scumpia PO, Delano MJ, Weinstein JS, Cuenca AG, Wynn JL, et al. Type I interferon signaling in hematopoietic cells is required for survival in mouse polymicrobial sepsis by regulating CXCL10. The Journal of Experimental Medicine. 2010;207:319-26.
48. Piehler J, Thomas C, Garcia KC, Schreiber G. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunological Reviews. 2012;250:317-34.
49. Yamaoka K, Saharinen P, Pesu M, Holt VE, 3rd, Silvennoinen O, O’Shea JJ. The Janus kinases (Jaks). Genome Biol. 2004;5:253.
50. Casanova JL, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity. 2012;36:515-28.
51. Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokineinduced biologic responses. Cell. 1998;93:373-83.
52. Leonard WJ, O’Shea JJ. Jaks and STATs: biological implications. Annual Review of Immunology. 1998;16:293-322.
53. Wallweber HJ, Tam C, Franke Y, Starovasnik MA, Lupardus PJ. Structural basis of recognition of interferonalpha receptor by tyrosine kinase 2. Nature Structural & Molecular Biology. 2014;21:443-8.
54. Lupardus PJ, Ultsch M, Wallweber H, Bir Kohli P, Johnson AR, Eigenbrot C. Structure of the pseudokinasekinase domains from protein kinase TYK2 reveals a mechanism for Janus kinase (JAK) autoinhibition. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:8025-30.
55. Ungureanu D, Wu J, Pekkala T, Niranjan Y, Young C, Jensen ON, et al. The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nature Structural & Molecular Biology. 2011;18:971-6.
56. Shan Y, Gnanasambandan K, Ungureanu D, Kim ET, Hammaren H, Yamashita K, et al. Molecular basis for pseudokinase-dependent autoinhibition of JAK2 tyrosine kinase. Nature Structural & Molecular Biology. 2014;21:579-84.
57. Ferrao R, Wallweber HJ, Ho H, Tam C, Franke Y, Quinn J, et al. The Structural Basis for Class II Cytokine Receptor Recognition by JAK1. Structure. 2016;24:897- 905.
58. Jiang J, Zhao M, Chang C, Wu H, Lu Q. Type I Interferons in the Pathogenesis and Treatment of Autoimmune Diseases. Clin Rev Allergy Immunol. 2020;59:248-72.
59. Wahren-Herlenius M, Dorner T. Immunopathogenic mechanisms of systemic autoimmune disease. Lancet. 2013;382:819-31.
60. Decker T, Kovarik P, Meinke A. GAS elements: a few nucleotides with a major impact on cytokine-induced gene expression. Journal of Interferon & Cytokine Research : the Official Journal of the International Society for Interferon and Cytokine Research. 1997;17:121-34.
61. Darnell JE, Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415-21.
62. Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nature Immunology. 2017;18:374-84.
63. Chen X, Bhandari R, Vinkemeier U, Van Den Akker F, Darnell JE, Jr., Kuriyan J. A reinterpretation of the dimerization interface of the N-terminal domains of STATs. Protein Sci. 2003;12:361-5.
64. Mao X, Ren Z, Parker GN, Sondermann H, Pastorello MA, Wang W, et al. Structural bases of unphosphorylated STAT1 association and receptor binding. Molecular cell. 2005;17:761-71.
65. Neculai D, Neculai AM, Verrier S, Straub K, Klumpp K, Pfitzner E, et al. Structure of the unphosphorylated STAT5a dimer. The Journal of Biological Chemistry. 2005;280:40782-7.
66. Tsai MH, Pai LM, Lee CK. Fine-Tuning of Type I Interferon Response by STAT3. Frontiers in Immunology. 2019;10:1448.
67. Delgoffe GM, Vignali DA. STAT heterodimers in immunity: A mixed message or a unique signal? JAKSTAT. 2013;2:e23060.
68. Ho HH, Ivashkiv LB. Role of STAT3 in type I interferon responses. Negative regulation of STAT1- dependent inflammatory gene activation. The Journal of Biological Chemistry. 2006;281:14111-8.
69. Tyler DR, Persky ME, Matthews LA, Chan S, Farrar JD. Pre-assembly of STAT4 with the human IFN-alpha/ beta receptor-2 subunit is mediated by the STAT4 N-domain. Mol Immunol. 2007;44:1864-72.
70. Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, Melero I. Direct effects of type I interferons on cells of the immune system. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:2619-27.
71. Lekmine F, Sassano A, Uddin S, Majchrzak B, Miura O, Druker BJ, et al. The CrkL adapter protein is required for type I interferon-dependent gene transcription and activation of the small G-protein Rap1. Biochemical and Biophysical Research Communications. 2002;291:744-50.
72. Hsu YA, Huang CC, Kung YJ, Lin HJ, Chang CY, Lee KR, et al. The anti-proliferative effects of type I IFN involve STAT6-mediated regulation of SP1 and BCL6. Cancer Letters. 2016;375:303-12.
73. Gupta S, Jiang M, Pernis AB. IFN-alpha activates Stat6 and leads to the formation of Stat2:Stat6 complexes in B cells. Journal of Immunology. 1999;163:3834-41.